![str1]()
AM 2394
1-(6′-(2-hydroxy-2-methylpropoxy)-4-((5-methylpyridin-3-yl)oxy)-[3,3′-bipyridin]-6-yl)-3-methylurea
Urea, N-[6′-(2-hydroxy-2-methylpropoxy)-4-[(5-methyl-3-pyridinyl)oxy][3,3′-bipyridin]-6-yl]-N‘-methyl-
CAS 1442684-77-6
Chemical Formula: C22H25N5O4
Exact Mass: 423.1907
Array Biopharma Inc., Amgen Inc. INNOVATORS
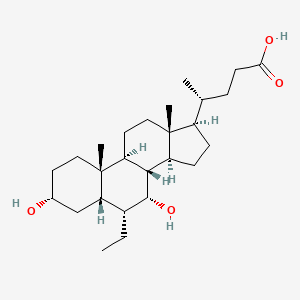
AM-2394 is a potent and selective Glucokinase agonist (GKA), which catalyzes the phosphorylation of glucose to glucose-6-phosphate. AM-2394 activates GK with an EC50 of 60 nM, increases the affinity of GK for glucose by approximately 10-fold, exhibits moderate clearance and good oral bioavailability in multiple animal models, and lowers glucose excursion following an oral glucose tolerance test in an ob/ob mouse model of diabetes
Type 2 diabetes mellitus (T2DM) is a disease characterized by elevated plasma glucose in the presence of insulin resistance and inadequate insulin secretion. Glucokinase (GK), a member of the hexokinase enzyme family, catalyzes the phosphorylation of glucose to glucose-6-phosphate in the presence of ATP.
![img]()
![str1]()
Glucokioase i exok ase IV or D> is a glycolytic enssyiris that plays, an importaat. role irt blood sugar regulation .related to glucose utifeattoti a»d metabolism in the liver and pancreatic beta •cells. Serving as a glucose sessor, gtoeokiuase controls lasma glucose, levels. Glucokinaae plays a doal rob in .reducing plasma glucose levels; glucose-mediated activation of the en¾ymc in hepatocytes facilitates hepatic giocose npiafcc aad glycogen synthesis, while that la pancreatic beta ceils ultimately induces ins lin seeretio«. Both of these effects in turn reduce plasma glucose levels.
Clinical evidence has shown that, glueokitiase variants with, decreased, and increased activities are associated with mature easel, diabetes of the y ung { O0Y2) and persistent: hyperinsul nemic hypoglycemia &( infancy (PHHI), respectively. lso, aoo n.sulin dependent diabetes rneilitos (NIDDM) patients have been reported to have inappropriately lo giueokaiase activity; Ftirtherrnare. overexpressioa of glucokiuase it* dietary or gesetie animal models of diabetes either prevents, aoKiiorafes, or reverses the progress of pathological. symptoms in the disease. For these reasons, compounds that activate gfecokiaase have been sought by the pitasaaceatjeai liidustry.
International patent application, Publication No. WO 2 7/OS3345, which was published on May 10, 200?, discloses as giocokinase act ators certain 2-an«.aopyridiiie derivatives bearing at the 3 -position a meihyieneoxy-dkrked aromatic group a d on. the ammo group a heteroaryl ring, such as dna/oly! or i A4-lmadiazoiyl
it has .now been found that pyridyl ureas are useful as glneokirtase activators. Cettain of these •compounds have been, found to have an outstanding combination of properties that especially adapts them, for oral use to control plasma glucose levels.
![]()
![]()
Novel Series of Potent Glucokinase Activators Leading to the Discovery of AM-2394
† Departments of Therapeutic Discovery, Metabolic Disorders, and Pharmacokinetics and Drug Metabolism, Amgen Inc., 1120 Veterans Boulevard, South San Francisco, California 94080, United States
‡ Departments of Metabolic Disorders, Comparative Biology and Safety Sciences and Pharmacokinetics and Drug Metabolism, Amgen Inc., One Amgen Center Drive, Thousand Oaks, California 91320, United States
§ Array BioPharma Inc., 3200 Walnut Street, Boulder, Colorado 80301, United States
ACS Med. Chem. Lett., Article ASAP
DOI: 10.1021/acsmedchemlett.6b00140
http://pubs.acs.org/doi/abs/10.1021/acsmedchemlett.6b00140
![Abstract Image]()
Glucokinase (GK) catalyzes the phosphorylation of glucose to glucose-6-phosphate. We present the structure–activity relationships leading to the discovery of AM-2394, a structurally distinct GKA. AM-2394 activates GK with an EC50 of 60 nM, increases the affinity of GK for glucose by approximately 10-fold, exhibits moderate clearance and good oral bioavailability in multiple animal models, and lowers glucose excursion following an oral glucose tolerance test in an ob/ob mouse model of diabetes.
![]()
![]()
PATENT
WO 2013086397
COPYING ERROR
Example. 1734 t¾^Jtiyi¾rea
Step A: In 100 mL of DMA were corafeiaed 1 ^545miSO- -ll«omp ridinr2-yl)-3-i«e hir8a- (17.5 g, 70,5 ii!-!to!). 5-o:ieS:t}yI yiidlii~3- ). (9,24 g, S4.7 ΪΗΪΪΪΟ!}, sad CO · (10.1 g, 77.6 mmo!) mid heated to 90 *C for 5 days. After that time, the reaction was om lete a d to it was added water arid DCM and stirred vigorously for 3 hr. The resulting solid was isolated via vacuum .filtratiott nd the cake was wasted mill rater and DCM. The DCM in tli aqueous rime was dried vdth a stream of aidogeji aad vigorous sbrriug. Use resulting solid was then collected via vacuum filtration aad these solids were
Stirred vig rousl in f 0% MeOH irt EtOAc arid die res dtipg solid was colleeied. via vactiiars fiirfati m.
Trie two batches wen i coiiibiaed to yield I-(5-bmmo-4 5^»ie†fey pyiidin-3-yl xy)p Tidin-2- d 3~ metbySurea (I S J g, 5 3.7 om»)i, 76% yield).
S e .8: In 2 niL ofc ioxane
yI) iyridMJ-2-yios:y)pf¾ps3i-2-oI (0,098 g, 0.33 «ΜΠΟΪ), “ -i5-bs¾tao-4-{5-a3fidiy I py f idia-3 – ylosy)f5yridia-2-yl)-3-raethyl«rea (0.075 g, 0.22 tn ol.. t, and.2M poiass.ua» carbonate (0.33 ml, 0.67 m oi} artd tfets was s parged wi h At .for 10 mia before PdC§4dppl)*DCM (0.01 g g, 0.022 msttol) was added and dre reae!io a was sparged for aaotber 5 ma-, ir efore a was sealed and heated to 100 oversight The react! art was then loaded directly onto s ilica gel (50% acetone to PCM w4i. }%
MH40H) to afford i – (6′-(2diydioxy-2i-H5eth:ylpropCis:y) -4-{ 5″i:t re th y Ipy r i d i rt -3- io s y ) -3 ,3 : -bipyr id i rt -6- yl)-3-aie5¾ylt)rea φ.? 42 , 0.096 m ol, 43 % yield). !1 1 HMR (400 Mife, CDCij) 3 ppm 9.06 is,. !H),
S.33 is, 1H>, 8,27 (rs 2H), 8. Π (s, I H): K. (s, IHU 82 (dd, j-S.fi, 5.9 H HI), 1.21 (S !H), 6,«8
(d, Hz, i i i ). 6. ,4 (s:. m>, 4.25 (s, 2H), 2,87 (dj =4,3 Hz„ 3H) 2,37 (s, 3H>. 1 .33 is, <SH). Mass speetram (apci) tar/, : – 423.9 (M÷H).
REFERENCES
Novel Series of Potent Glucokinase Activators Leading to the Discovery of AM-2394
Paul J. Dransfield, Vatee Pattaropong, Sujen Lai, Zice Fu, Todd J. Kohn, Xiaohui Du, Alan Cheng, Yumei Xiong, Renee Komorowski, Lixia Jin, Marion Conn, Eric Tien, Walter E. DeWolf Jr., Ronald J. Hinklin, Thomas D. Aicher, Christopher F. Kraser, Steven A. Boyd, Walter C. Voegtli, Kevin R. Condroski, Murielle Veniant-Ellison, Julio C. Medina, Jonathan Houze, and Peter Coward
Publication Date (Web): May 23, 2016 (Letter)
DOI: 10.1021/acsmedchemlett.6b00140
/////////Glucokinase activator, GKA, AM-2394, 1442684-77-6, AM 2394, Amgen
O=C(NC)NC1=CC(OC2=CC(C)=CN=C2)=C(C3=CC=C(OCC(C)(O)C)N=C3)C=N1
Filed under:
Uncategorized Tagged:
1442684-77-6,
AM-2394,
amgen,
GKA,
Glucokinase Activator ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()






































































 Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim talked in his keynote…
Dr Friedrich Haefele, Vice President Fill & Finish Biopharma at Boehringer Ingelheim talked in his keynote…






















































































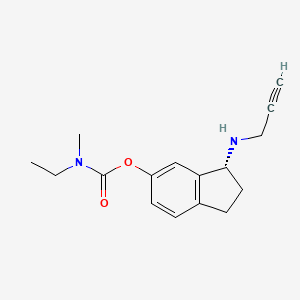





 (R)-3-(Prop-2-ynylamino)-2,3-dihydro-1H-inden-5-yl ethyl(methyl)carbamate
(R)-3-(Prop-2-ynylamino)-2,3-dihydro-1H-inden-5-yl ethyl(methyl)carbamate

 (R)-3-(Prop-2-ynylamino)-2,3-dihydro-1H-inden-5-yl ethyl(methyl)carbamate hemi((2R,3R)-2,3-dihydroxysuccinate)
(R)-3-(Prop-2-ynylamino)-2,3-dihydro-1H-inden-5-yl ethyl(methyl)carbamate hemi((2R,3R)-2,3-dihydroxysuccinate)