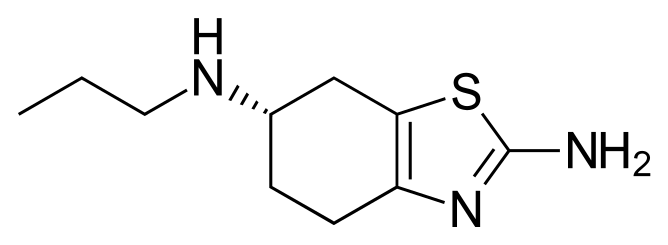
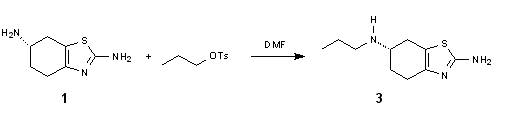
![]()
![Figure imgf000045_0001]()
![]()
Bexagliflozin
THR1442; THR-1442, EGT 0001442; EGT1442
CAS :1118567-05-7
(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2- (cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol
1-[4-Chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-1-deoxy-beta-D-glucopyranose
1,5-Anhydro-1(S)-[4-chloro-3-[4-[2-(cyclopropyloxy)ethoxy]benzyl]phenyl]-D-glucitol
Chemical Formula: C24H29ClO7
Exact Mass: 464.16018Mechanism of Action:SGLT2 inhibitor
Indication:Type 2 diabetes
Development Stage:Phase II
Developer:Theracos, Inc.
![]()
![Figure US20130267694A1-20131010-C00062]() DIPROLINE COMPLEX
DIPROLINE COMPLEX
![]()
Bexagliflozin diproline
RN: 1118567-48-8, C24-H29-Cl-O7.2C5-H9-N-O2
Molecular Weight, 695.2013
![im1]()
![]()
Bexagliflozin [(2S,3R,4R,5S,6R)-2-[4-chloro-3-({4-[2-(cyclopropyloxy) ethoxy] phenyl} methyl)phenyl]-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol] is an orally administered drug for the treatment of Type 2 Diabetes Mellitus (T2DM) and is classified as a Sodium Glucose co-Transporter 2 (SGLT2) Inhibitor. It is in Phase 2b study to evaluate the effect of bexagliflozin tablets in subjects with type 2 diabetes mellitus.

Bexagliflozin, also known as EGT1442, is a potent and selective SGLT2 inhibitor, attenuates blood glucose and HbA(1c) levels in db/db mice and prolongs the survival of stroke-prone rats. The IC(50) values for EGT1442 against human SGLT1 and SGLT2 are 5.6μM and 2nM, respectively. In normal rats and dogs a saturable urinary glucose excretion was produced with an ED(50) of 0.38 and 0.09mg/kg, respectively. EGT1442 showed favorable properties both in vitro and in vivo and could be beneficial to the management of type 2 diabetic patients.
One promising target for therapeutic intervention in diabetes and related disorders is the glucose transport system of the kidneys. Cellular glucose transport is conducted by either facilitative (“passive”) glucose transporters (GLUTs) or sodium-dependent (“active”) glucose cotransporters (SGLTs). SGLTl is found predominantly in the intestinal brush border, while SGLT2 is localized in the renal proximal tubule and is reportedly responsible for the majority of glucose reuptake by the kidneys. Recent studies suggest that inhibition of renal SGLT may be a useful approach to treating hyperglycemia by increasing the amount of glucose excreted in the urine (Arakawa K, et al., Br J Pharmacol 132:578-86, 2001; Oku A, et al., Diabetes 48:1794-1800, 1999).
![]()
The potential of this therapeutic approach is further supported by recent findings that mutations in the SGL T2 gene occur in cases of familial renal glucosuria, an apparently benign syndrome characterized by urinary glucose excretion in the presence of normal serum glucose levels and the absence of general renal dysfunction or other disease (Santer R, et al., J Am Soc Nephrol 14:2873-82, 2003). Therefore, compounds which inhibit SGLT, particularly SGL T2, are promising candidates for use as antidiabetic drugs.
Compounds previously described as useful for inhibiting SGLT include C-glycoside derivatives (such as those described in US6414126, US20040138439, US20050209166, US20050233988, WO2005085237, US7094763, US20060009400, US20060019948, US20060035841, US20060122126, US20060234953, WO2006108842, US20070049537 and WO2007136116), O-glycoside derivatives (such as those described in US6683056, US20050187168, US20060166899, US20060234954, US20060247179 and US20070185197), spiroketal-glycoside derivatives (described in WO2006080421), cyclohexane derivatives (such as those described in WO2006011469), and thio- glucopyranoside derivatives (such as those described in US20050209309 and WO2006073197).
![]()
PATENT
WO 2009026537……………PRODUCT PATENT
http://www.google.co.in/patents/WO2009026537A1?cl=en
![]()
![]()
Example 19
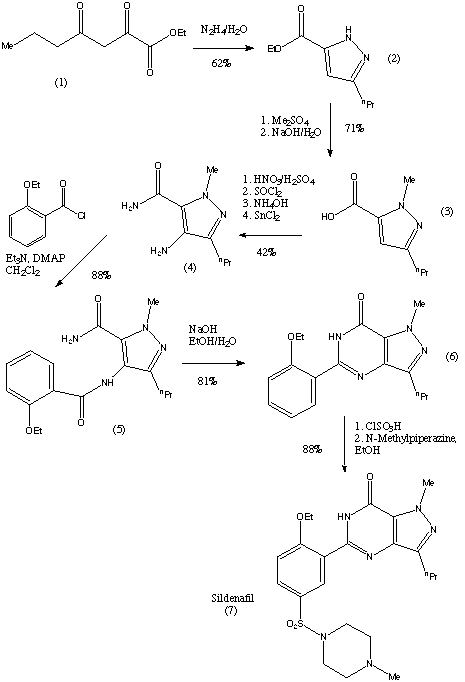
[0289] The synthesis of compound BQ within the invention is given below.
[0290] Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (0.87 g, 36.1 mmol) and iodine (catalytic) in THF (4 mL) was added slowly BrCH2CH2Br (4.6 g, 24.5 mmol) in THF (8 mL). The exothermic reaction was cooled in an ice-bath. After complete addition OfBrCH2CH2Br, a solution of 2- (2-bromoethyl)-l,3-dioxolane (1 g, 5.6 mmol) was added dropwise. The reaction mixture was then kept at reflux for 24 h, quenched by addition of aqueous NH4Cl, and extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give crude intermediate BO (400 mg) as yellow oil. [0292] Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
Ts0^°V
To a solution of 2-cyclopropoxyethanol (400 mg, 3.92 mmol) in DCM (10 niL) were added TsCl (821 mg, 4.31 mmol) and Et3N (0.6 mL, 4.31 mmol). The reaction was stirred at room temperature overnight. Then, IN HCl was added, and the reaction was extracted with DCM. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to give a yellow oil. The oil was purified by preparative TLC to obtain intermediate BP (50 mg) as a yellow oil.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2- cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Compound BQ)
To a solution of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (intermediate Dl) (30 mg, 0.08 mmol) in anhydrous DMF (1 mL) were added 2-cyclopropoxyethyl 4-methylbenzenesulfonate (intermediate BP) (20 mg, 0.08 mmol) and Cs2CO3 (52 mg, 0.16 mmol). The mixture was stirred at room temperature for 12 h. Then the reaction mixture was poured into water, extracted with EA, washed with brine, dried with anhydrous Na2SO4 and concentrated to an oil. The oil was purified by preparative HPLC to obtain compound BQ (11 mg) as a colorless oil. 1H NMR (CD3OD): δ 7.30 (m, 3H), 7.11 (d, J= 8.8 Hz, 2H), 6.82 (d, J= 8.8 Hz, 2H), 4.13 (m, 5H), 3.85 (m, 3H), 3.81 (m, IH), 3.40 (m, 4H), 3.30 (m, IH), 0.52 (m, 4H); MS ESI (m/z) 465 (M+H)+, calc. 464.
![]()
Example 33
The synthesis of complex DM within the invention is outlined in FIG. 30, with the details given below.
Preparation of 2-cyclopropoxyethanol (Intermediate BO)
To a suspension of Mg powder (86.7 g, 3.6 mol) and I2 (catalytic) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) at a rate that maintained the reaction temperature between 40-55° C. A solution of 2-(2-bromoethyl)-1,3-dioxolane (100 g, 0.56 mol) in anhydrous THF (750 mL) was added dropwise, and the reaction mixture was kept at 40-55° C. for 16 h. The reaction was quenched by addition of an aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give intermediate BO (27 g) as yellow oil, which was used in the next step without further purification.
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (Intermediate BP)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added crude 2-cyclopropoxyethanol from the previous step (27 g, 0.26 mol) at −5 to 0° C. A solution of p-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise, and the reaction mixture was kept at −5 to 0° C. for 16 h. The reaction mixture was then incubated at room temperature for 30 min, the organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4 and concentrated to get the crude intermediate BP as a yellow oil (53.3 g), which was used for the preparation of intermediate DK below without further purification.
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (Intermediate H)
To a stirred solution of 4-bromo-1-chloro-2-(4-ethoxybenzyl)benzene (intermediate B) (747 g, 2.31 mol) in dichloromethane was added slowly boron tribromide (1.15 kg, 4.62 mol) at −78° C. The reaction mixture was allowed to warm to room temperature. When the reaction was complete as measured by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with an aqueous solution of saturated sodium bicarbonate, then with water, and then with brine, and dried over Na2SO4. The residue was concentrated and then recrystallized in petroleum ether to obtain intermediate H as a white solid (460 g, yield 68%). 1H NMR (CDCl3, 400 MHz): δ 7.23˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Preparation of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (Intermediate DK)
A mixture of 4-(5-bromo-2-chlorobenzyl)phenol (56.7 g, 210 mmol) and Cs2CO3 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 30 min, and then 2-cyclopropoxyethyl 4-methylbenzenesulfonate (crude intermediate BP from the second preceeding step above) (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight, and then diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, then with brine, and dried over Na2SO4. The residue was concentrated and then purified by flash column chromatography on silica gel (eluent PE:EA=10:1) to give intermediate DK as a liquid (51 g, yield 64%). 1H NMR (CDCl3, 400 MHz): δ 7.22˜7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52 (m, 2H).
Preparation of (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (Intermediate DL)
![Figure US08802637-20140812-C00113]()
To a stirred solution of 4-bromo-1-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (213 g) in anhydrous THF/toluene (1:2 v/v, 1.7 L) under argon was added n-BuLi (2.5 M in hexane, 245.9 mL) dropwise at −60±5° C. The mixture was stirred for 30 min, and then transferred to a stirred solution of (3R,4S,5R,6R)-3,4,5-tris(trimethylsilyloxy)-6-((trimethylsilyloxy)methyl)tetrahydro-2H-pyran-2-one (310.5 g) in toluene (1.6 L) at −60±5° C. The reaction mixture was continuously stirred at −60±5° C. for 1 before quenching with an aqueous solution of saturated ammonium chloride (1.5 L). The mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 mL). The combined organic layers were washed with brine (1 L), dried over Na2SO4, and concentrated. The residue was dissolved in methanol (450 mL), and methanesulfonic acid (9.2 mL) was added at 0° C. The solution was allowed to warm to room temperature and stirred for 2.0 h. The reaction was quenched with an aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and then additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The crude product was used in the next step without further purification.
Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex (Complex DM)
![Figure US08802637-20140812-C00114]()
To a stirred solution of crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol from the previous step in CH2Cl2/CH3CN (1:1, 1.3 L) at −5° C. was added triethylsilane (28.2 mL, 563 mmol), followed by BF3.Et2O (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm gradually to room temperature. The reaction was quenched by addition of an aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2SO4 and concentrated to give the crude product (230 g, purity 82.3%). To the crude product was added L-proline (113.7 g) in EtOH/H2O (15:1 v/v, 2.09 L), and the mixture was stirred at 80° C. for 1 h until it became a clear solution. Hexane (3.0 L) was added dropwise over 50 min, while the temperature was maintained at about 60° C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/H2O (15:1 v/v, 2×300 mL), hexane (2×900 mL), and dried at 45° C. under vacuum for 10 h to give pure complex DM as a white solid (209 g; HPLC purity 99.2% (UV)). 1H NMR (CD3OD, 400 MHz): δ 7.25˜7.34 (m, 3H), 7.11 (d, J=8.8 Hz, 2H), 6.84 (d, J=8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53 (m, 2H).
Crystalline complex DM was analyzed by X-ray powder diffraction using CuKα1 radiation. The diffraction pattern is shown inFIG. 31 and summarized in Table 1 (only peaks up to 30° in 2θ are listed). The melting point of complex DM was determined by differential scanning calorimetry (DSC) as 151±1° C. (evaluated as onset-temperature; heating from 50° C. to 200° C. at 10° C./min). The DSC spectrum is shown in FIG. 32.
Preparation of (3R,4R,5S,6R)-2-(4-chloro-3-(4-hydroxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate D)
To a stirred solution of (3R,4R,5S,6R)-2-(4-chloro-3-(4-ethoxybenzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (Intermediate C) (2 g, 5.9 mmol) in dichloromethane was added BBr3 (14.6 mL, 1 M) dropwise at −78° C. After the addition was complete, the mixture was allowed to warm to 0° C. and held at this temperature for 2 h. When LC-MS showed that no starting material remained, the mixture was cooled to −78° C. again, and quenched with water. When the temperature was stable, saturated NaHCO3 solution was added. The mixture was evaporated under reduced pressure, and the residue was extracted with EtOAc. The organic layer was washed with NaHCO3 and brine, dried over Na2SO4, evaporated and purified to obtain intermediate D (0.7 g).
In addition, for use in the synthesis of certain compounds of the invention, the 2S isomer (intermediate D1) and the 2R isomer (intermediate D2) of intermediate D were separated by preparative LC-MS. Intermediate D1: 1H NMR (CD3OD): δ 7.30 (m, 3H), 6.97 (d, 2H, J=6.8 Hz), 6.68 (d, 2H, J=6.8 Hz), 4.56 (s, 1H), 4.16 (s, 1H), 3.91˜4.02 (m, 5H), 3.79 (m, 1H), 3.64 (m, 1H). Intermediate D2: 1H NMR (CD3OD): δ 7.29˜7.33 (m, 3H), 7.00 (d, 2H, J=6.8 Hz), 6.70 (d, 2H, J=6.8 Hz), 4.58 (d, 1H, J=4.0 Hz), 3.96˜4.02 (m, 4H), 3.93˜3.95 (m, 1H), 3.81˜3.85 (m, 1H), 3.64˜3.69 (m, 1H).
PATENT
http://www.google.com/patents/US20130267694
![]()
Example 14 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol crystals
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(442-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol bis(L-proline) complex in methanol/water solvent mixture.
![Figure US20130267694A1-20131010-C00066]()
(2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (1.3 kg) was added to a propylene drum (25 L) and methanol (3.6 kg) and water (1.3 kg) and the mixture was stirred until the solids dissolved. The solution was filtered through filter membrane (Millipore, 0.45 μm) into a clean glass reactor (50 L). The mixture was refluxed for 30 min and water (7.2 kg) was added over 1.0 h while maintaining the temperature between 50 and 65° C. The mixture was slowly cooled to ˜42° C. over 2 h. A suspension of seed crystal (26 g) in cold (−5° C.) mixture of methanol/water (78 mL, 2.8/6.5 (w/w)) and the slow cooling was continued to −5° C. over 12 h. The suspension was stirred for another 5 h and was filtered. The solid was slurried with cold water and filtered (0 to 5° C., 3×2.6 kg). The filter cake was dried under reduced pressure for 24 h until the loss on drying was no more than 0.5% to give a white solid (825 g, 92% yield, 99.3% pure by \HPLC-0001).
Example 15 Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
This example describes preparation of 4-(2-chloro-5-iodobenzyl)phenol using gaseous hydrobromic acid.
Preparation of (2-chloro-5-iodophenyl)methan-1-ol
![Figure US20130267694A1-20131010-C00068]()
A 250 mL of 4-necked flask equipped with thermometer and mechanical stirring was charged with NaBH4 (4.16 g, 0.11 mol) and THF (60 mL) under argon. After cooling to 0˜5° C. with stirring, a solution of iodine in THF (12.7 g I2 in 25 mL THF) was added slowly dropwise over 30 min and the reaction temperature was maintained below 10° C. After the addition was completed, a solution of 2-chloro-5-iodobenzoic acid (15.0 g, 50 mmol) in THF (20 mL) was added dropwise over 30 min and kept the reaction temperature below 10° C. After stirring for another 3 h at 20˜25° C., the reaction mixture was heated to reflux for additional 16 h and monitored by TLC (PE/EA=1:1, Rf=0.2). The mixture was cooled to 20˜25° C. and poured into ice water (100 mL), extracted with ethyl acetate (2×100 mL), washed with water (2×100 mL), brine (100 mL), concentrated and the residue was purified by flash chromatography (PE:EA=20:1 as eluant, 200 mL) to give an off-white solid. Yield: 10.0 g (70%) MS ESI (m/z): 269 [M+1]+.
Preparation of 4-(2-Chloro-5-Iodobenzyl)Phenol
![Figure US20130267694A1-20131010-C00069]()
A 100 mL of 4-necked flask equipped with thermometer and mechanical stirrer was charged with (2-chloro-5-iodophenyl)methanol (268.5 mg, 1 mmol), anhydrous ZnCl2 (136.3 mg, 1 mmol), dichloromethane (5.0 mL) and n-hexane (29 mL) under argon. After stirring for 10 min at 20 to 25° C., HBr (gas) was bubbled into the mixture for 10 min and a solution of phenol (197.6 mg, 2.1 mmol) in dry dichloromethane (3.0 mL) was added dropwise over 30 min. After bubbling HBr for additional 2 h, the mixture was refluxed for 3 days. The conversion was about 65%. The mixture was quenched with ice water (50 mL), extracted with ethyl acetate (2×30 mL), washed with water (2×30 mL), brine (30 mL), concentrated and the residue was purified by flash chromatography (PE:EA=25:1 as eluant, 200 mL) to give an off-white solid. Yield: 180 mg (52%). 1H NMR (CDCl3, 400 MHz): δ 7.44 (d, J=8.4 Hz, 2H), 7.03˜7.09 (m, 3H), 6.77 (d, J=8.4 Hz, 2H), 4.76 (s, 1H), 3.95 (s, 2H), 3.82 (s, 2H). MS ESI (m/z): 345 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 154.1, 141.4, 139.5, 136.6, 134.2, 131.2, 130.9, 130.1, 115.5, 91.67, 38.07.
Example 16 Preparation of 2-(4-(2-Cyclopropoxyethoxy)Benzyl)-1-Chloro-4-Iodobenzene
This example describes the preparation of 2-(4-(2-cyclopropoxyethoxy)benzyl)-1-chloro-4-iodobenzene via coupling of the 4-(2-chloro-5-iodobenzyl)phenol with 2-cyclopropoxyethyl 4-methylbenzenesulfonate.
Under nitrogen a 500 L glass-lined reactor was charged with acetone (123 kg) with stirring (120 RPM), 4-(2-chloro-5-iodobenzyl)phenol (19.37 kg, 0.056 kmol), 2-cyclopropoxyethyl 4-methylbenzenesulfonate (15.85 kg, 0.062 kmol), cesium carbonate (18.31 kg, 0.0562 kmol) powder, potassium carbonate (23.3 kg, 0.169 kmol) powder and TBAI (4.15 kg, 0.011 kmol). After stirring for 4045 h at 40° C., TLC (PE:EA=4:1, Rf=0.3) showed that starting material was consumed. The mixture was cooled to 20˜25° C.
The reaction mixture was filtered over diatomite (28 kg) and the filter cake was washed with acetone (2×31 kg). The combined filtrates were transferred to a 500 L glass-lined reactor and concentrated. The residue was dissolved in ethyl acetate (175 kg, washed with water (2×97 kg) and concentrated until the volume was about 100 L and was transferred to a 200 L glass-lined reactor and continued to concentrate to get about 22.5 kg of crude material.
The crude material was dissolved in methanol/n-hexane (10:1, 110 kg) under refluxing for 30 min with stirring (100 RPM) until it was a clear solution. The mixture was cooled to 5 to 10° C. and some crystal seeds (20 g) were added. The suspension was stirred for another 5 h at 5 to 10° C. The mixture was filtered at 0 to 5° C. and the filter cake was washed with pre-cooled methanol/n-hexane (10:1, 5° C., 2×11 kg). The filter cake was dried under at 15 to 20° C. for 15 h to give off-white to white solid. Yield: 18.1 kg, 75%. Melting Point: 31° C. (DSC onset). 1H NMR (CDCl3, 400 MHz): δ 7.45˜7.50 (m, 2H), 7.09˜7.12 (m, 3H), 6.88 (d, J=8.8 Hz, 2H), 4.11 (t, J=5.2 Hz, 2H), 3.99 (s, 2H), 3.88 (t, J=5.2 Hz, 2H), 3.40˜3.44 (m, 1H), 0.63˜0.67 (m, 2H), 0.49˜0.54 (m, 1H). MS ESI (m/z): 429 [M+1]+. 13C NMR (CDCl3, 100 MHz): δ 157.5, 141.5, 139.5, 136.6, 134.2, 131.2, 130.8, 129.9, 114.9, 91.66, 69.00, 67.13, 53.72, 38.08, 5.63.
Example 9 Preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(L-proline) complex by co-crystallization of ((2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol with L-proline in ethanol/water/n-heptane solvent mixture.
![Figure US20130267694A1-20131010-C00062]()
The crude (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol (2.5 kg) was added to a glass reactor containing ethanol (95%, 16 kg) and L-proline (1.24 kg) and the mixture was refluxed for 1 h. While keeping the temperature above 60° C., n-heptane (8.5 kg) was added over 40 min. The mixture was slowly cooled to 25 to 20° C. and stirred at this temperature for 10 h. The mixture was filtered and the solids were washed with cold (−5° C.) ethanol (95%, 2×2.5 L) and n-heptane (2×5 L) and the solids were dried under reduced pressure at 55 to 65° C. for 20 h to give a white solid (3.03 kg, 81% yield, 99.4% pure by HPLC-0001).
Example 7 Preparation of ((2S,3R,4R,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)Tetrahydro-2H-Pyran-3,4,5-triol
This example describes preparation of (2S,3R,4R,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol by removal of the anomeric OH or OMe.
(2S,3R,4S,5S,6R)-2-(4-Chloro-3-(4-(2-Cyclopropoxyethoxy)Benzyl)Phenyl)-6-(Hydroxymethyl)-2-Methoxytetrahydro-2H-Pyran-3,4,5-Triol Solution
A 30 L glass reactor equipped with a thermometer was charged with crude (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol (1.15 kg), DCM (2.3 kg) and acetonitrile (1.4 kg), and the mixture was magnetically stirred until all the solids dissolved under nitrogen sparging. The solution was cooled to ˜−15° C.
Triethylsilane Solution:
BF3.Et2O (1.2 kg) was added to a cold (−20 to −15° C.) solution of triethysilane (1.08 kg) dichloromethane (2.3 kg) and acetonitrile (1.4 kg) with nitrogen sparging.
The cold (2S,3R,4S,5S,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy)benzyl)phenyl)-6-(hydroxymethyl)-2-methoxytetrahydro-2H-pyran-3,4,5-triol solution was added to the cold triethylsilane solution at such a rate to maintain the temperature between −20 and −15° C. (˜2 to 3 h).
The reaction mixture was stirred for another 2 to 3 h and then quenched by addition of an aqueous solution of sodium bicarbonate (7.4% w/w, 7.8 kg) and the reaction mixture was stirred for about 15 min. The solvents were removed under reduced pressure (2 h, temperature below 40° C.). The residue was partitioned between ethyl acetate (6.9 kg) and water (3.9 kg). The layers were separated and the aqueous layer was extracted with ethyl acetate (2×3.5 kg). The combined organic layers were washed with brine (2×3.8 kg) and the solvents were removed under reduced pressure. Anhydrous ethanol (2.3 kg) was added and concentrated to give the crude product of the title compound (1 kg, 90% yield, 90% HPLC-0001) as yellow solid.
PATENT
WO 2011153953
https://www.google.com/patents/WO2011153953A1?cl=en
Example 1. Preparation of (2S.iR. R.5S.6R)-2-(4-chloro-3-(4-(2-cvclopropoxyethoxy) benzyl)phenyl)-6-(hvdroxymethyl)tetrahvdro-2H-pyran-3,4,5-triol, bis(X-proline) complex
Example 1A
Preparation of 2-cyclopropoxyethanol (1)
To a suspension of Mg powder (86.7 g, 3.6 mol) and iodine (cat) in anhydrous THF (0.7 L) was added slowly 1,2-dibromoethane (460 g, 2.4 mol) in anhydrous THF (2 L) slowly at a rate as to keep the internal temperature between 40-55 °C. After the addition, a solution of 2-(2-bromoethyl)-l,3-dioxolane (lOOg, 0.56 mol) in anhydrous THF (750 mL) was added dropwise. The reaction mixture was kept at 40-55 °C for 16h and was quenched by addition of aqueous solution of ammonium chloride. The mixture was extracted with methylene chloride. The organic layer was dried over sodium sulfate, and concentrated to give the title product (27 g) as yellow oil, which was directly used without further purification.
Example IB
Preparation of 2-cyclopropoxyethyl 4-methylbenzenesulfonate (2)
To a stirred solution of sodium hydroxide (32 g, 0.8 mol) in water (180 mL) and THF (180 mL) was added Example 1A (27 g, 0.26 mol) at -5 to 0 °C. Afterwards, a solution of ji?-toluenesulfonyl chloride (52 g, 0.27 mol) in THF (360 mL) was added dropwise. The reaction mixture was kept at -5 to 0 °C for 16 h. The reaction mixture was then kept at room temperature for 30 min. The organic layer was separated and the aqueous layer was extracted with ethyl acetate (2×1.0 L). The combined organic layers were washed with brine, dried over Na2S04 and concentrated to get the crude product as yellow oil (53.3 g). It was used directly without further purification.
Example 1C
Preparation of 4-(5-bromo-2-chlorobenzyl)phenol (3)
To a stirred solution of 4-bromo-l-chloro-2-(4-ethoxybenzyl)benzene (747 g, 2.31 mol) in dichloromethane was added boron tribromide (1.15 kg, 4.62 mol) slowly at -78 °C. The reaction mixture was allowed to rise to room temperature. When the reaction was complete as measure by TLC, the reaction was quenched with water. The mixture was extracted with dichloromethane. The organic layer was washed with aqueous solution of saturated sodium bicarbonate, water, brine, dried over Na2S04, and concentrated. The residue was recrystallized in petroleum ether to give the title compound as a white solid (460 g, yield 68%). 1H NMR (CDC13, 400MHz): δ 7.23-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.79 (d, J=8.8 Hz, 2H), 5.01 (s, 1H), 4.00 (s, 2H).
Example ID
Preparation of 4-bro -l-chloro-2-(4-(2-cyclopropoxyethoxy)benzyl)benzene (4)
A mixture of Example 1C (56.7 g, 210 mmol) and Cs2C03 (135 g, 420 mmol) in DMF (350 mL) was stirred at room temperature for 0.5 h. Example IB (53.3 g, 210 mmol) was added. The reaction mixture was stirred at room temperature overnight. It was diluted with water (3 L) and extracted with EtOAc. The organic layer was washed with water, brine, dried over Na2S04, and concentrated. The residue was purified by flash column
chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1) to give the title compound as liquid (51 g, yield 64%). 1H NMR (CDC13, 400MHz): δ 7.22-7.29 (m, 3H), 7.08 (d, J=8.8 Hz, 2H), 6.88 (d, J=8.8 Hz, 2H), 4.10 (t, J=4.8 Hz, 2H), 3.86 (t, J=4.8 Hz, 2H), 3.38-3.32 (m, 1H), 0.62-0.66 (m, 2H), 0.49-0.52(m, 2H).
Example IE
Preparation of (25,5R, S,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6-(hydroxymethyl)-2-metlioxytetraliydro-2H-pyran-3,4,5-triol (5)
![Figure imgf000034_0002]()
To a stirred solution of Example ID (213 g) in anhydrous THF/toluene (1 :2 (v/v), 1.7 L) under argon was added n-BuLi (2.5 M hexane, 245.9 mL) drop wise at -60 ± 5 °C. The mixture was stirred for 30 min. before transferred to a stirred solution of 2,3,4,6-tetra-O- trimethylsilyl-P-Z -glucolactone (310.5 g) in toluene (1.6 L) at -60 ± 5 °C. The reaction mixture was continuously stirred at -60 ± 5 °C for 1 h before quenching with aqueous solution of saturated ammonium chloride (1.5 L). Then mixture was allowed to warm to room temperature and stirred for 1 h. The organic layer was separated and the water layer was extracted with ethyl acetate (3×500 niL). The combined organic layers were washed with brine (1 L), dried over Na2S04, and concentrated. The residue was dissolved in methanol (450 mL) and methanesulfonic acid (9.2 mL) was added at 0 °C. The solution was allowed to warm to room temperature and stirred for 20 h. It was quenched with aqueous solution of sodium bicarbonate (50 g) in water (500 mL) and additional water (900 mL) was added. The mixture was extracted with ethyl acetate (3×1.0 L). The combined organic layers were washed with brine, dried over Na2S04, concentrated and used directly in the next step without further purification.
Example IF
Preparation of (25,5R, R,55,6R)-2-(4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H-pyran-3,4,5-triol, bis(Z-proline) complex (7)
![Figure imgf000035_0001]()
To stirred solution of Example IE in CH2C12/CH3CN (650 mL:650 mL) at -5 °C was added triethylsilane (28.2 mL, 563 mmol), and followed by BF3-Et20 (52.3 mL, 418.9 mmol). The reaction was stirred for 16 h while the temperature was allowed to warm to room temperature gradually. The reaction was quenched with aqueous solution of saturated sodium bicarbonate to pH 8.0. The organic volatiles were removed under vacuum. The residue was partitioned between ethyl acetate (2.25 L) and water (2.25 L). The organic layer was separated, washed with brine, dried over Na2S04 and concentrated to give the crude product 6 (230 g, purity 82.3%). This product and L-proline (113.7 g) in EtOH/H20 (15:1 v/v, 2.09 L) was stirred at 80 °C for 1 h when it became a clear solution. Hexane (3.0 L) was added dropwise into the above hot solution over 50 min, with the temperature being kept at about 60 °C. The reaction mixture was stirred overnight at room temperature. The solid was filtered and washed with EtOH/ H20 (15:1 (v/v), 2×300 mL), hexane (2×900 mL), and dried at 45 °C under vacuum for 10 h to give the pure title compound 7 as a white solid (209 g).
Purity (HPLC) 99.2% (UV). 1H NMR (CD3OD, 400 MHz): δ 7.25—7.34 (m, 3H), 7.11 (d, J = 8.8 Hz, 2H), 6.84 (d, J= 8.8 Hz, 2H), 4.03-4.11 (m, 5H), 3.96-4.00 (m, 2H), 3.83-3.90 (m, 3H), 3.68-3.72 (m, 1H), 3.36-3.46 (m, 6H), 3.21-3.30 (m, 3H), 2.26-2.34 (m, 2H), 2.08-2.17 (m, 2H), 1.94-2.02 (m, 4H), 0.56-0.57 (m, 2H), 0.52-0.53(m, 2H).
Example 2. Direct Preparation of Crystalline Compound 8 from Complex 7
This example illustrates the preparation of a crystalline form of (2S, 3R, 4R, 5S, 6R)-2- (4-chloro-3-(4-(2-cyclopropoxyethoxy) benzyl)phenyl)-6- (hydroxymethyl)tetrahydro-2H- pyran-3,4,5-triol.
![Figure imgf000036_0001]()
To a 5.0 L 4-necked flask equipped with a mechanical stirrer was added the starting co-crystal (150.0 g) and methanol (300 mL). The mixture was stirred at room temperature with mechanical stirring (anchor agitator, 2-blades 9 cm) until a cloudy solution/suspension formed, to which distilled water (1500 mL) was added dropwise at a rate of -12.5 mL/min. As the mixture warmed from the exotherm of adding water to methanol, the mixture became clear after adding about 1/5 to 1/3 of the water. After the addition was completed the reaction was stirred continuously at 80 rpm for another 5 h. The reaction mixture was filtered over medium-speed filter paper and the filter cake was washed with distilled water (450 mL and then 300 mL) and dried under vacuum using an oil pump (~6 mm Hg) at 45 °C for 48 hours to give the target product as a white crystalline solid (94.2 g, 93.9% yield, purity (HPLC): 99.3%).
Example 5. Indirect Preparation of Crystalline Compound 8 from Complex 7
![Figure imgf000038_0001]()
[0113] To a 200 L glass lined reactor equipped with a double-tier paddle agitator and a glass condenser was added sequentially complex 7 (7.33 kg), ethyl acetate (67.5 kg) and pure water (74.0 kg). The mixture was heated to reflux and stirred at reflux for 30 min. The reaction mixture was cooled to approximately 50 °C and the organic layer was separated and the aqueous layer was extracted with ethyl acetate (34.0 kg). The combined organic layers were washed with pure water (3×74.0 kg) (IPC test showed that the IPC criteria for L-proline residue was met after three water washes). The mixture was concentrated at 40 °C under vacuum (-15 mmHg) for 3 h until the liquid level dropped below the lower-tier agitator paddle. The mixture (18 kg) was discharged and transferred to a 20L rotary evaporator. The mixture was concentrated under vacuum (40 °C, ~5 mmHg) to a minimum volume. The remaining trace amount of ethyl acetate was removed azeotropically at 40 °C under vacuum with methanol (10 kg). The residue was dried under vacuum of an oil pump (~6 mmHg) at 40 °C for 10 h to give 8 as a white amorphous solid (4.67 kg, purity (HPLC): 99.2%) which was used in the next step without further purification.
The recrystallization was accomplished by the following steps. To a 100 L glass line reactor equipped with a double-tier paddle agitator and a glass condenser was added the above amorphous 8 (4.67 kg) and methanol (18.0 kg). The mixture was refluxed at 70 °C for 30 min until a clear solution formed, to which pure water (45.0 kg) was added over 2 hours. After the addition was completed (the reaction temperature was 41 °C), the reaction mixture was cooled to room temperature and stirred at room temperature for 15 hours. The reaction mixture was filtered and the wet cake was washed with pure water (2×15 kg) and dried under vacuum at 55-60 °C for 12 hours to give the target product as an off-white crystalline solid (3.93 kg, yield: 84% in two steps; purity (HPLC): 99.7%).
Example 6. Direct Preparation of Crystalline Compound 8 from Amorphous 8
A 5 L 4-neck flask was charged with 8 (amorphous), 116 g, and methanol (580 mL). The reaction mixture was heated to 60 C with mechanical stirring and the solution became clear. Water (2320 mL) was added dropwise to the reaction solution at 40 mL/min at 50 °C. The reaction mixture was stirred overnight at room temperature. The reaction mixture was filtered and the filter cake was washed with water (2×200 mL), dried under vacuum at 55 °C for 12 hours, to afford white crystalline 8. Yield is 112.8 g (97.2%).
References:
1. Clinical Trial, A Dose Range Finding Study to Evaluate the Effect of Bexagliflozin Tablets in Subjects With Type 2 Diabetes Mellitus. NCT02390050 (retrieved on 26-03-2015).
| WO2008144346A2 * |
May 15, 2008 |
Nov 27, 2008 |
Squibb Bristol Myers Co |
Crystal structures of sglt2 inhibitors and processes for their preparation |
| WO2009026537A1 * |
Aug 22, 2008 |
Feb 26, 2009 |
Theracos Inc |
Benzylbenzene derivatives and methods of use |
| CN1407990A * |
Oct 2, 2000 |
Apr 2, 2003 |
布里斯托尔-迈尔斯斯奎布公司 |
C-aryl glucoside sgltz inhibitors
| WO2008144346A2 * |
May 15, 2008 |
Nov 27, 2008 |
Squibb Bristol Myers Co |
Crystal structures of sglt2 inhibitors and processes for their preparation |
| WO2009026537A1 * |
Aug 22, 2008 |
Feb 26, 2009 |
Theracos Inc |
Benzylbenzene derivatives and methods of use |
| CN1407990A * |
Oct 2, 2000 |
Apr 2, 2003 |
布里斯托尔-迈尔斯斯奎布公司 |
C-aryl glucoside sgltz inhibitors |
|
| WO2010022313A2 * |
Aug 21, 2009 |
Feb 25, 2010 |
Theracos, Inc. |
Processes for the preparation of sglt2 inhibitors |
////////
Filed under:
DIABETES,
Phase2 drugs,
Phase3 drugs,
Uncategorized Tagged:
bexagliflozin,
DIABETES,
flozin,
Inc.,
PHASE 3,
Theracos ![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()




















 Originally posted on
Originally posted on 







































































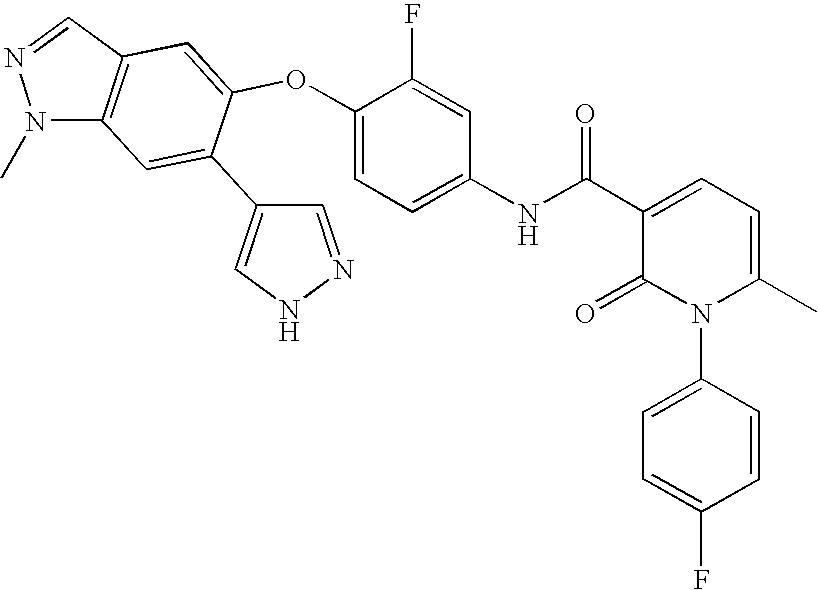



 1 MERESTENIB
1 MERESTENIB




























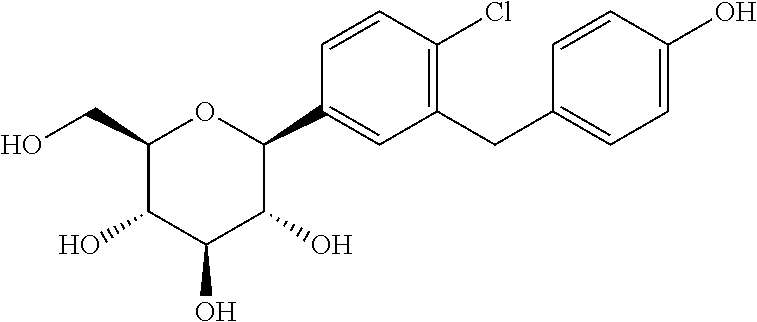







































































 BMS 248360 A DIFFERENT COMPD
BMS 248360 A DIFFERENT COMPD





































 Originally posted on
Originally posted on 























 LEVITRA
LEVITRA









































 Methane sulfonate
Methane sulfonate