
Mercaptamine bitartrate
2-aminoethanethiol;2,3-dihydroxybutanedioic acid
| Molecular Formula: | C6H13NO6S |
|---|---|
| Molecular Weight: | 227.231 g/mol |
Cystagon; Cysteamine – Mylan/Orphan Europe; Cysteamine bitartrate
Procysbi; CYSTEAMINE BITARTRATE; 27761-19-9; CHEBI:50386; (+/-)-Tartaric Acid
| INGREDIENT | UNII | CAS | |
|---|---|---|---|
| Cysteamine Bitartrate | QO84GZ3TST | 27761-19-9 | |
| Cysteamine Hydrochloride | IF1B771SVB | 156-57-0 |
Cysteamine bitartrate is a mercaptoethylamine compound that is endogenously derived from the COENZYME A degradative pathway. The fact that cysteamine is readily transported into LYSOSOMES where it reacts with CYSTINE to form cysteine-cysteamine disulfide and CYSTEINE has led to its use in CYSTINE DEPLETING AGENTS for the treatment of CYSTINOSIS.
Cysteamine Bitartrate is an aminothiol salt used in the treatment of nephropathic cystinosis. Cysteamine bitartrate enters the cell and reacts with cystine producing cysteineand cysteine–cysteamine mixed disulfide compound, both of which, unlike cystine, can pass through the lysosomal membrane. This prevents the accumulation of cystinecrystals in the lysosomes of patients with cystinosis, which can cause considerable damage and eventual destruction of the cells, particularly in the kidneys. (NCI05)
Cysteamine is a simple aminothiol molecule that is used to treat nephropathic cystinosis, due to its ability to decrease the markedly elevated and toxic levels of intracellular cystine that occur in this disease and cause its major complications. Cysteamine has been associated with serum enzyme elevations when given intravenously in high doses, but it has not been shown to cause clinically apparent acute liver injury.
Given intravenously or orally to treat radiation sickness. The bitartrate salts (Cystagon® and Procysbi) have been used for the oral treatment of nephropathic cystinosis and cystinurea. The hydrochloride salt (Cystaran ) is indicated for the treatment of corneal cystine crystal accumulation in cystinosis patients.
) is indicated for the treatment of corneal cystine crystal accumulation in cystinosis patients.
- OriginatorMylan
- DeveloperAlphapharm; Mylan
- ClassMercaptoethylamines; Small molecules; Sulfhydryl compounds
- Mechanism of ActionGlutathione synthase stimulants
Highest Development Phases
- MarketedNephropathic cystinosis
- DiscontinuedUnspecified
Most Recent Events
- 09 Apr 2018Mercaptamine bitartrate licensed to Recordati worldwide
- 26 Oct 2017Chemical structure information added
- 31 Dec 2008Mercaptamine bitartrate oral is still in phase II/III trials for Undefined indication in European Union
DESCRIPTION: CYSTAGON® (cysteamine bitartrate) Capsules for oral administration, contain cysteamine bitartrate, a cystine depleting agent which lowers the cystine content of cells in patients with cystinosis, an inherited defect of lysosomal transport. CYSTAGON® is the bitartrate salt of cysteamine, an aminothiol, beta-mercaptoethylamine. Cysteamine bitartrate is a highly water soluble white powder with a molecular weight of 227 and the molecular formula C2H7NS · C4H6O6. It has the following chemical structure:

Cysteamine is a medication intended for a number of indications, and approved by the FDA to treat cystinosis.
It is stable aminothiol, i.e., an organic compound containing both an amine and a thiol functional groups. Cysteamine is a white, water-soluble solid. It is often used as salts of the ammonium derivative [HSCH2CH2NH3]+[1] including the hydrochloride, phosphocysteamine, and bitartrate.[2]
Cysteamine molecule is biosynthesized in mammals, including humans, by the degradation of coenzyme A. The intermedia pantetheineis broken down into cysteamine and pantothenic acid.[2] It is the biosynthetic precursor to the neurotransmitter hypotaurine.[3][4]
Medical uses
Cysteamine is used to treat cystinosis. It is available by mouth (capsule and extended release capsule) and in eye drops.[5][6][7][8][9]
Adverse effects
Topical use
The most important adverse effect related to topical use might be skin irritation.
Oral use
The label for oral formulations of cysteamine carry warnings about symptoms similar to Ehlers-Danlos syndrome, severe skin rashes, ulcers or bleeding in the stomach and intestines, central nervous symptoms including seizures, lethargy, somnolence, depression, and encephalopathy, low white blood cell levels, elevated alkaline phosphatase, and idiopathic intracranial hypertension that can cause headache, tinnitus, dizziness, nausea, double or blurry vision, loss of vision, and pain behind the eye or pain with eye movement.[6]
The main side effects are Ehlers-Danlos syndrome, severe skin rashes, ulcers or bleeding in the stomach and intestines, central nervous symptoms, low white blood cell levels, elevated alkaline phosphatase, and idiopathic intracranial hypertension (IIH). IIH can cause headache, ringing in the ears, dizziness, nausea, blurry vision, loss of vision, and pain behind the eye or with eye movement.
Additional adverse effects of oral cysteamine include bad breath, skin odor, vomiting, nausea, stomach pain, diarrhea, and loss of appetite.[6]
The drug is in pregnancy category C; the risks of cysteamine to a fetus are not known but it harms babies in animal models at doses less than those given to people.[7][8]
For eye drops, the most common adverse effects are sensitivity to light, redness, and eye pain, headache, and visual field defects.[8]
Interactions
There are no drug interactions for normal capsules or eye drops,[7][8] but the extended release capsules should not be taken with drugs that affect stomach acid like proton pump inhibitors or with alcohol, as they can cause the drug to be released too quickly.[6] It doesn’t inhibit any cytochrome P450 enzymes.[6]
Pharmacology
People with cystinosis lack a functioning transporter (cystinosin) which transports cystine from the lysosome to the cytosol. This ultimately leads to buildup of cystine in lysosomes, where it crystallizes and damages cells.[5] Cysteamine enters lysosomes and converts cystine into cysteine and cysteine-cysteamine mixed disulfide, both of which can exit the lysosome.[6]
Biological function
Cysteamine also promotes the transport of L-cysteine into cells, that can be further used to synthesize glutathione, which is one of the most potent intracellular antioxidants.[4]
Cysteamine is used as a drug for the treatment of cystinosis; it removes cystine that builds up in cells of people with the disease.[10]
History
First evidence regarding the therapeutic effect of cysteamine on cystinosis dates back to 1950s. Cysteamine was first approved as a drug for cystinosis in the US in 1994.[6] An extended release form was approved in 2013.[11]
Society and culture
It is approved by FDA and EMA.[5][6]
In 2013, the regular capsule of cysteamine cost about $8,000 per year; the extended release form that was introduced that year was priced at $250,000 per year.[11]
Research
It was studied in in vitro and animal models for radiation protection in the 1950s, and in similar models from the 1970s onwards for sickle cell anemia, effects on growth, its ability to modulate the immune system, and as a possible inhibitor of HIV.[2]
In the 1970s it was tested in clinical trials for Paracetamol toxicity which it failed, and in clinical trials for systemic lupus erythematosus in the 1990s and early 2000s, which it also failed.[2]
Clinical trials in Huntington’s disease were begun in the 1990s and were ongoing as of 2015.[2][12]
As of 2013 it was in clinical trials for Parkinson’s disease, malaria, radiation sickness, neurodegenerative disorders, neuropsychiatric disorders, and cancer treatment.[10][2]
It has been studied in clinical trials for pediatric nonalcoholic fatty liver disease[13]
Horizon Pharma , following the acquisition of Raptor Pharmaceuticals (previously through its Bennu Pharmaceuticals subsidiary, and following its acquisition of Encode Pharmaceuticals , which licensed the drug from the University of California )) has developed and launched DR Cysteamine (EC Cysteamine; Procysbi), a methyl-CpG binding protein 2 (MECP2) gene modulating, oral delayed-release (DR), enteric-coated (EC), bitartrate salt formulation of mercaptamine (cysteamine).
PRODUCT PATENT, WO2007089670 ,
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2007089670
hold SPC protection in most of the EU states until September 2028, and expire in the US in July 2037. In July 2018, the US FDA’s Orange Book was seen to list a patent covering product ( US8026284 and US9173851 ) of cysteamine bitartrate, that is due to expire in September 2027 and December 2034, respectively.
Cystinosis is a rare, autosomal recessive disease caused by intra-lysosomal accumulation of the amino acid cystine within various tissues, including the spleen, liver, lymph nodes, kidney, bone marrow, and eyes. Nephropathic cystinosis is associated with kidney failure that
necessitates kidney transplantation. To date, the only specific treatment for nephropathic cystinosis is the sulfhydryl agent, cysteamine. Cysteamine has been shown to lower intracellular cystine levels, thereby reducing the rate of progression of kidney failure in children.
[0004] Cysteamine, through a mechanism of increased gastrin and gastric acid production, is ulcerogenic. When administered orally to children with cystinosis, cysteamine has also been shown to cause a 3 -fold increase in gastric acid production and a 50% rise of serum gastrin levels. As a consequence, subjects that use cysteamine suffer
gastrointestinal (GI) symptoms and are often unable to take cysteamine regularly or at full dose .
[0005] To achieve sustained reduction of leukocyte cystine levels, patients are normally required to take oral cysteamine every 6 hours, which invariably means having to awaken from sleep. However, when a single dose of
cysteamine was administered intravenously the leukocyte cystine level remained suppressed for more than 24 hours, possibly because plasma cysteamine concentrations were higher and achieved more rapidly than when the drug is administered orally. Regular intravenous administration of cysteamine would not be practical. Accordingly, there is a need for formulations and delivery methods that would result in higher plasma, and thus intracellular, concentration as well as decrease the number of daily doses and therefore improve the quality of life for patients.
PATENT
US-20180193292
Process for the preparation of cysteamine bitartrate . Represents the first patenting to be seen from Lupin Limited on cysteamine bitartrate.
|
Cysteamine bitartrate (I) is a cystine depleting agent which lower the cystine content of cells in patients with cystinosis, an inherited defect of lysosomal transport, it is indicated for the management of nephropathic cystinosis in children and adults. Cysteamine bitartrate (I) is simplest stable aminothiol salt and has the following structural formula:
|
|
The application WO 2014204881 provides pharmaceutical composition of cysteamine bitrate and another application WO 2007089670 provides method of administrating cysteamine and pharmaceutically salts and method of treatment thereof.
|
Examples
1. Preparation of Cysteamine Bitartrate.
|
A mixture of ethanol (1000 ml), butylated hydroxy anisole (1 g) and cysteamine hydrochloride (100 g) was stirred and cooled to 5 to 10° C. To this mixture a solution of ethanol (500 ml) and sodium hydroxide (352 g) was added over a period of 30 minutes.
|
|
The mixture was stirred at a temperature of 10 to 15° C. for 45 minutes. The mixture was filtered through celite. The filtrate was added to a mixture of ethanol (1250 ml), butylated hydroxy anisole (1 g) and L-(+)-tartaric acid (132 g) at a temperature of 55-60° C. The reaction mixture was stirred at 70-75° C. for 45 minutes. The mixture was cooled to 20-30° C. The solid was filtered, washed with ethanol and dried under vacuum.
|
2. Purification of Cysteamine Bitartrate.
|
A mixture of cysteamine bitartrate (100 g) and ethanol (5000 ml) was heated to a temperature of 77-82° C. The solution was filtered and the filtrate was cooled to 20 to 30° C. and stirred for 40 minutes. The solid was filtered, washed with ethanol and dried under vacuum. Yield: 80 g; HPLC purity: 99.90%.
|
3. Preparation of Crystalline Form L1 of Cysteamine Bitartrate.
|
A mixture of cysteamine bitartrate (50 g) and methanol (600 ml) was heated to a temperature of 35-45° C. The solution was filtered and the filtrate was cooled to 5 to 10° C. Cysteamine bitartrate (0.25 g) seed material was added to the filtrate. The slurry was cooled to −5 to −25° C. and stirred for 40 minutes. The solid was filtered, washed with precooled methanol and dried under vacuum. Yield: 40 g. Cysteamine bitartrate with X-ray powder diffraction pattern as depicted in FIG. 1 was obtained.
|
4. Preparation of Crystalline Form L2 of Cysteamine Bitartrate.
|
A mixture of cysteamine bitartrate (50 g), butylated hydroxy anisole (1.3 g) and methanol (600 ml) was heated to a temperature of 35-45° C. The solution was filtered and the filtrate was cooled to 5 to 10° C. Cysteamine bitartrate (0.25 g) seed material was added to the filtrate. The slurry was cooled to −25 to −30° C. and stirred for 40 minutes. The solid was filtered, washed with precooled methanol and the solid was dried under 800-900 mm/Hg of vacuum at 35-40° C. for 5 hours. Yield: 40 g. Cysteamine bitartrate with X-ray powder diffraction pattern as depicted in FIG. 2 was obtained.
|
PATENT
WO 2014204881
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2014204881
References
- Jump up^ Reid, E. Emmet (1958). Organic Chemistry of Bivalent Sulfur. 1. New York: Chemical Publishing Company, Inc. pp. 398–399.
- ^ Jump up to:a b c d e f Besouw, M; Masereeuw, R; van den Heuvel, L; Levtchenko, E (August 2013). “Cysteamine: an old drug with new potential”. Drug Discovery Today. 18 (15–16): 785–92. doi:10.1016/j.drudis.2013.02.003. PMID 23416144.
- Jump up^ Singer, Thomas P (1975). “Oxidative Metabolism of Cysteine and Cystine”. In Greenberg, David M. Metabolic pathways Vol. 7. Metabolism of sulfur compounds (3rd ed.). New York: Academic Press. p. 545. ISBN 9780323162081.
- ^ Jump up to:a b Besouw, Martine; Masereeuw, Rosalinde; van den Heuvel, Lambert; Levtchenko, Elena (August 2013). “Cysteamine: an old drug with new potential”. Drug Discovery Today. 18(15–16): 785–792. doi:10.1016/j.drudis.2013.02.003. ISSN 1878-5832. PMID 23416144.
- ^ Jump up to:a b c Nesterova, Galina; Gahl, William A. (October 6, 2016). “Cystinosis”. GeneReviews. University of Washington, Seattle.
- ^ Jump up to:a b c d e f g h “US Label: Cysteamine bitartrate delayed-release capsules” (PDF). FDA. August 2015.
- ^ Jump up to:a b c “US Label: Cysteamine bitartrate capsules” (PDF). FDA. June 2007.
- ^ Jump up to:a b c d “US Label: Cysteamine ophthalmic solution” (PDF). FDA. October 2012.
- Jump up^ Shams, F; Livingstone, I; Oladiwura, D; Ramaesh, K (10 October 2014). “Treatment of corneal cystine crystal accumulation in patients with cystinosis”. Clinical ophthalmology (Auckland, N.Z.). 8: 2077–84. doi:10.2147/OPTH.S36626. PMC 4199850
 . PMID 25336909.
. PMID 25336909. - ^ Jump up to:a b Besouw, Martine; Masereeuw, Rosalinde; van den Heuvel, Lambert; Levtchenko, Elena (August 2013). “Cysteamine: an old drug with new potential”. Drug Discovery Today. 18(15–16): 785–792. doi:10.1016/j.drudis.2013.02.003. ISSN 1878-5832. PMID 23416144.
- ^ Jump up to:a b Pollack, Andrew (30 April 2013). “F.D.A. Approves Raptor Drug for Form of Cystinosis”. The New York Times.
- Jump up^ Shannon, KM; Fraint, A (15 September 2015). “Therapeutic advances in Huntington’s Disease”. Movement disorders : official journal of the Movement Disorder Society. 30 (11): 1539–46. doi:10.1002/mds.26331. PMID 26226924.
- Jump up^ Mitchel, EB; Lavine, JE (November 2014). “Review article: the management of paediatric nonalcoholic fatty liver disease”. Alimentary pharmacology & therapeutics. 40 (10): 1155–70. doi:10.1111/apt.12972. PMID 25267322.

 |
|
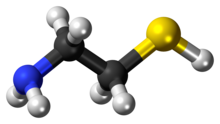
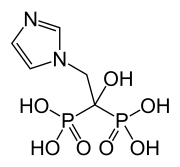
Skeletal formula (top)
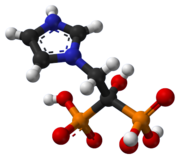
Ball-and-stick model of the cysteamine |
|
| Clinical data | |
|---|---|
| Synonyms | 2-Aminoethanethiol β-Mercaptoethylamine 2-Mercaptoethylamine Decarboxycysteine Thioethanolamine Mercaptamine |
| License data | |
| Identifiers | |
| CAS Number | |
| PubChemCID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| ECHA InfoCard | 100.000.421  |
| Chemical and physical data | |
| Formula | C2H7NS |
| Molar mass | 77.15 g·mol−1 |
| Melting point | 95 to 97 °C (203 to 207 °F) |

///////////Mercaptamine bitartrate, Cystagon, Cysteamine, Cysteamine bitartrate, Mercaptamine,, システアミン , меркаптамин , 巯乙胺
C(CS)N.C(C(C(=O)O)O)(C(=O)O)O



























































 EP 0582304; JP 1994192195
EP 0582304; JP 1994192195 Chem Pharm Bull 1998,46(4),616
Chem Pharm Bull 1998,46(4),616







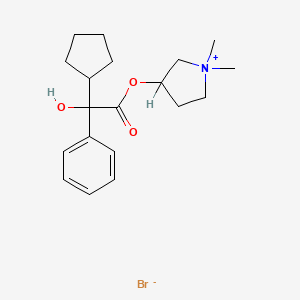

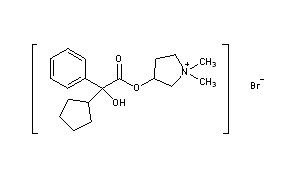











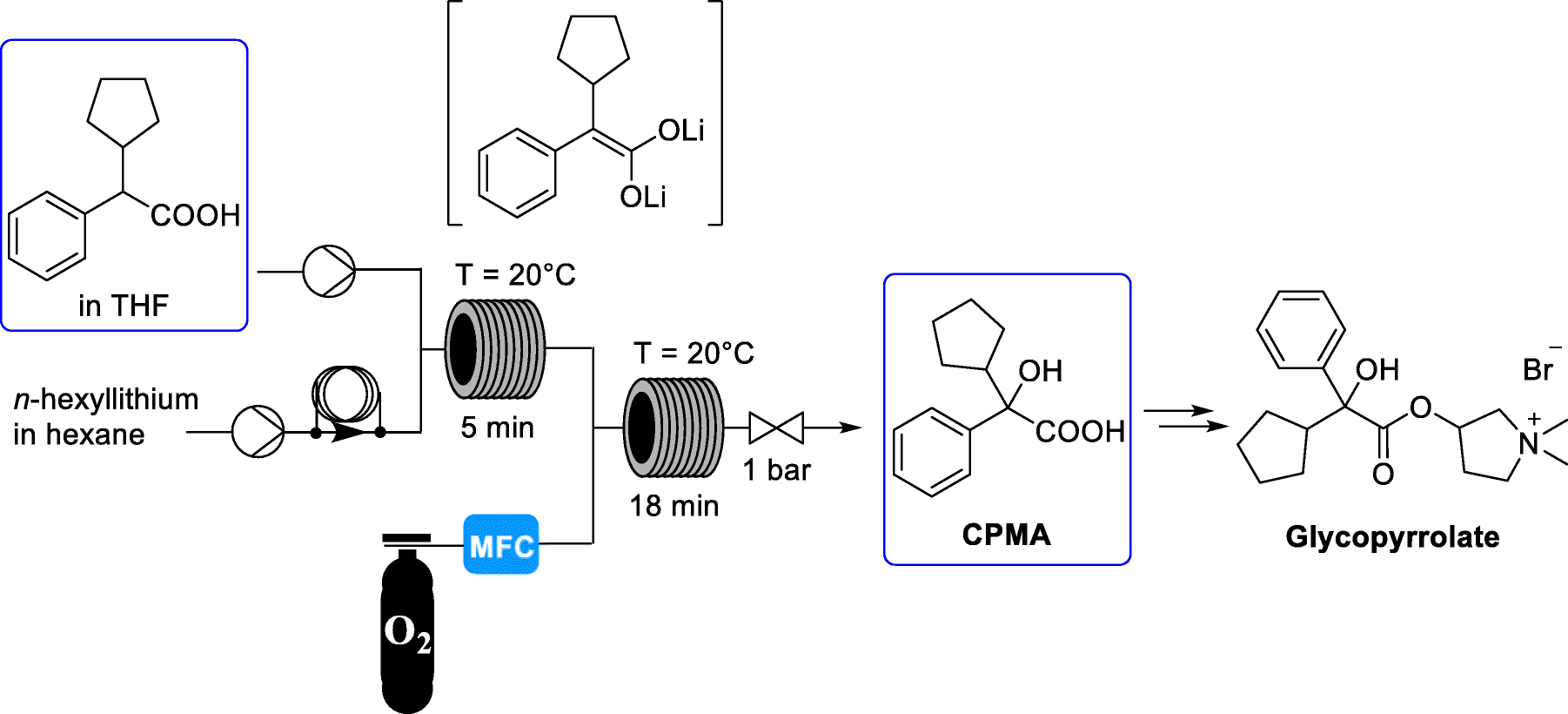


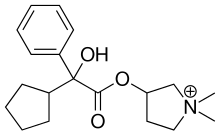




 C6H6O3S : 547.07
C6H6O3S : 547.07




















 Diazoxide
Diazoxide