GS-443902 is a bioactive ATP analogue with broad-spectrum antiviral activity [5] and is the same compound formed by remdesivir, though by a different enzymatic pathway. Unlike remdesivir, which is metabolized by enzymes that are highly expressed in the liver, GS-441524 released by obeldesivir is metabolized by enzymes that are evenly expressed throughout the body. Due to their different metabolic pathways, obeldesivir can be administered orally, whereas remdesivir must be administered intravenously for COVID-19 treatment.[citation needed]
The pharmacokinetic properties of obeldesivir and improved was first published by Chinese researchers in May 2022. The Chinese group pursued investigation of obeldesivir independently from Gilead Sciences. Compared to IV administered GS-441524 in rats at 5 mg/kg, orally administered obeldesivir at 25 mg/kg (referred to as “ATV006”) yielded approximately 22% bioavailability.[6] Treatment with obdeldesivir reduced viral load and prevents lung pathology in KI-hACE2 and Ad5-hACE2 mouse models of SARS-CoV-2. A patent filed by Gilead Sciences with a priority date of August 27, 2020,[7] found the bioavailability of GS-441524 after oral administration of obdeldesivir (compound 15) in mice, rats, ferrets, dogs, and cynomolgus macaques to be 41%, 63.9%, 154%, 94%, and 38%, respectively. Across all species evaluated, obeldesivir showed improved oral bioavailability compared to oral administration of the parent nucleoside, GS-441524
Remdesivir 1 is an phosphoramidate prodrug that releases the monophosphate of nucleoside GS-441524 (2) into lung cells, thereby forming the bioactive triphosphate 2-NTP. 2-NTP, an analog of ATP, inhibits the SARS-CoV-2 RNA-dependent RNA polymerase replication and transcription of viral RNA. Strong clinical results for 1 have prompted interest in oral approaches to generate 2-NTP. Here, we describe the discovery of a 5′-isobutyryl ester prodrug of 2 (GS-5245, Obeldesivir, 3) that has low cellular cytotoxicity and 3–7-fold improved oral delivery of 2 in monkeys. Prodrug 3 is cleaved presystemically to provide high systemic exposures of 2 that overcome its less efficient metabolism to 2-NTP, leading to strong SARS-CoV-2 antiviral efficacy in an African green monkey infection model. Exposure-based SARS-CoV-2 efficacy relationships resulted in an estimated clinical dose of 350–400 mg twice daily. Importantly, all SARS-CoV-2 variants remain susceptible to 2, which supports development of 3 as a promising COVID-19 treatment.
Synthesis of ((2R,3S,4R,5R)-5-(4-Aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methyl Isobutyrate (3)
To a solution of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile 4 (2000 mg, 6.0 mmol) (30) and isobutyric acid (638 mg, 7.2 mmol) in DMF (5 mL), N,N′-diisopropylcarbodiimide (914 mg, 7.2 mmol) was slowly added followed by 4-(dimethylamino)pyridine (737 mg, 6.0 mmol). The reaction mixture was stirred for 4 h and then diluted with ethyl acetate, washed with water and brine, dried over sodium sulfate, and concentrated under reduced pressure. The residue was subjected to silica gel chromatography, eluting with 20% MeOH in CH2Cl2 to provide the intermediate ((3aR,4R,6R,6aR)-6-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4-yl)methyl isobutyrate. LCMS m/z = 402.2 (M + 1). To a solution of the intermediate (1500 mg) in THF (10 mL), conc. HCl (2 mL) was added, and the mixture was stirred at rt for 4 h. The reaction mixture was diluted with CH2Cl2, washed with water, saturated aqueous bicarbonate, and brine, dried over sodium sulfate, concentrated, and subjected to silica gel chromatography, eluting with 30% MeOH in CH2Cl2 to afford the title compound (660 mg, 50%). 1H NMR (400 MHz, MeOH-d4) δ 7.88 (s, 1H), 6.96–6.85 (m, 2H), 4.50–4.27 (m, 4H), 4.16 (dd, J = 6.2, 5.3 Hz, 1H), 2.56 (p, J = 7.0 Hz, 1H), 1.14 (dd, J = 7.0, 3.8 Hz, 6H). LCMS m/z: 362.1 (M + 1). 13C NMR (400 MHz, CHCl3–d3) δ 175.9, 155.6, 147.9, 123.5, 110.2, 100.8, 116.9, 116.6, 81.3, 79.0, 74.0, 70.2, 62.9, 33.2, 18.7, 18.6. HRMS m/z: 362.14615, C16H20N5O5 362.14644.
Example 1. (3aR, 4R, 6R, 6aR)-4-(4-aminopyrrole[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl- Synthesis of 2,2-dimethyltetrahydrofuran[3,4-d][1,3]dioxolyl-4-carbonitrile (compound 5)
Dissolve 5.62g of compound 69-0 in 30mL of acetone, then add 11.50mL of 2,2-dimethoxypropane and 1.34mL of 98% sulfuric acid, stir at 45°C for half an hour, cool to room temperature, and remove by rotary evaporation Organic solvents. Extract with 100 mL of ethyl acetate and 100 mL of saturated sodium bicarbonate solution. Repeat the extraction three times. Combine the ethyl acetate layers, add anhydrous sodium sulfate to dry, and filter to remove sodium sulfate. The organic solvent was removed by rotary evaporation, and 6.20 g of compound 5 (white solid, yield 97%) was obtained through column chromatography (eluent: petroleum ether/ethyl acetate (v/v) = 1/2). The hydrogen spectrum of the obtained compound 5 was detected, and the results are as follows:
Example 2. ((3aR,4R,6R,6aR)-6-(4-aminopyrrole[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2 ,Synthesis of 2-dimethyltetrahydrofuran[3,4-d][1,3]dioxol-4-yl)methylisobutyrate (compound INT-1)
Dissolve 1.50g of compound 5 in 15ml of dichloromethane, then add 0.42mL of isobutyric acid and 55.40mg of 4-dimethylaminopyridine, stir for 10 minutes, add 1.02g of dicyclohexylcarbodiimide, and stir at room temperature. Stir for 24h. After column chromatography separation (eluent: petroleum ether/ethyl acetate (v/v) = 1/1), 1.71 g of compound INT-1 (white solid, yield 94%) was obtained. The hydrogen spectrum and carbon spectrum of the obtained compound INT-1 were detected, and the results are as follows:
Hydrogen spectrum: 1 H NMR (400MHz, CDCl 3 ,ZQF-RD01-2)δ(ppm):7.99(s,1H),6.99(d,J=4.6Hz,1H),6.62(d,J=4.6Hz,1H),5.72(br,2H),5.49(d,J=6.8Hz,1H),4.93-4.90(dd,J=6.8Hz,4.3Hz,1H),4.61-4.58(q,J=4.4Hz,1H),4.44-4.26(m,2H),2.61-2.50(m,1H),1.77(s,3H),1.42(s,3H),1.17-1.14(q,J=3.8Hz,6H)。
Carbon spectrum: 13 C NMR (100MHz, CDCl 3 ,ZQF-RD01-2)δ(ppm):176.7,155.2,147.3,123.5,117.2,116.7,115.6,112.6,100.0,83.8,83.0,82.0,81.4,63.1,33.8,26.4,25.6,18.9。
To a solution of (3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile, Intermediate 1 (2000 mg, 6.0 mmol) (Siegel et. al. J. Med. Chem.2017, 60, 1648-1661) in tetrahydrofuran (THF) was added N,N-dimethylaminopyridine (DMAP) (0.03 eq). To the reaction mixture, isobutyric anhydride (1.1 eq) was added slowly. After the completion of the staring material, the reaction mixture was concentrated and purified by flash chromatography using 20% methanol in DCM as an eluant to give ((3aR,4R,6R,6aR)-6-(4- aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxol-4- yl)methyl isobutyrate, Intermediate 1a. LCMS: MS m/z: 402.2 (M+1).
Concentrated HCl (5 eq, 1mL) was added to a solution of Intermediate 1a (1000 mg) in acetonitrile (10 mL), which was then stirred at room temperature for 2 hours. LCMS showed the product formation. The reaction was stopped after 4 hours. The reaction mixture was diluted with ethyl acetate and quenched with saturated bicarbonate. The organic layer was separated, washed with brine, dried over sodium sulphate, and concentrated. The residue was purified by flash chromatography using 30% methanol DCM as an eluant. The collected fractions were concentrated to give Intermediate 2.1H NMR (400 MHz, Methanol-d4) δ 7.88 (s, 1H), 6.96 – 6.85 (m, 2H), 4.50 – 4.27 (m, 4H), 4.16 (dd, J = 6.2, 5.3 Hz, 1H), 2.56 (p, J = 7.0 Hz, 1H), 1.14 (dd, J = 7.0, 3.8 Hz, 6H); LCMS: MS m/z: 362.1 (M+1).
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The compound has been disclosed in patent WO2020020374A1. It is an enhancer of Zeste homolog 2 (EZH2) inhibitor, and can be used for preventing or treating EZH2-mediated diseases, including brain cancer, thyroid cancer, cardiac sarcoma, lung cancer, oral cancer, stomach cancer and various other cancers.
Example 4: Preparation of the Compound N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-3-(ethyl(tetrahydro-2H-pyran-4-yl)amino)-2-methyl-5-(trans-3-morpholinylcyclobutoxy)benzamide (4)
The title compound was prepared by referring to Example 1 with cyclobutanol replaced by cis-3-morpholinylcyclobutyl-1-ol. The cis-3-morpholinylcyclobutyl-1-ol was prepared as follows:
3-benzyloxy-cyclobutyl-1-one (3 g, 17 mmol, 1 eq) and morpholine (2.97 g, 34 mmol, 2 eq) were dissolved in DCE (30 mL). After stirring at room temperature for 0.5 h, the solution was added with NaBH(OAc)3 (7.23 g, 34 mmol, 2 eq) and stirred at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure to remove the solvent, adjusted to basicity with aqueous NaHCO3 solution, and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate, filtered, and concentrated under reduced pressure to give a crude product, which was purified by a phase preparative column to give 4-(3-(benzyloxy)cyclobutyl)morpholine (2.9 g). M/z (ES+), [M+H] +=248.
4-(3-(benzyloxy)cyclobutyl)morpholine (2.9 g, 117.4 mmol) and methane sulfonic acid (20 mL) was added to dichloromethane (40 mL). The mixture was stirred overnight at room temperature and concentrated under reduced pressure to remove the solvent to give a crude product, which was purified by a preparative column to give cis-3-morpholinylcyclobutyl-1-ol; m/z (ES+), [M+H] +=158. The trans-3-morpholinylcyclobutyl-1-ol can also be separated and obtained by this method.
Golcadomide is a modulator of the E3 ubiquitin ligase complex containing cereblon (CRL4-CRBN E3 ubiquitin ligase), with potential immunomodulating and antineoplastic activities. Upon administration, golcadomide specifically binds to cereblon (CRBN), thereby affecting the ubiquitin E3 ligase activity, and targeting certain substrate proteins for ubiquitination. This induces proteasome-mediated degradation of certain transcription factors, some of which are transcriptional repressors in T-cells. This leads to modulation of the immune system, including activation of T-lymphocytes, and downregulation of the activity of other proteins, some of which play key roles in the proliferation of certain cancer cell types. CRBN, the substrate recognition component of the CRL4-CRBN E3 ubiquitin ligase complex, plays a key role in the ubiquitination of certain proteins.
Example 1: Synthesis of (S)-2-(2,6-Dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dione (Compound 1)
(S)-2-(2,6-Dioxopiperidin-3-yl)-4-((2-fluoro-4-(hydroxymethyl)benzyl)amino)isoindoline-1,3-dione: A suspension of (S)-4-amino-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (5.00 g, 18.3 mmol) and 2-fluoro-4-(hydroxymethyl)benzaldehyde (2.82 g, 18.30 mmol) in 2:1 dioxane-MeOH (75 mL) was cooled to 0° C. and B 10H 14 (4.92 g, 40.3 mmol) was added in small portions over 5 minutes. The reaction flask was fitted with a septum and needle vent (pressure) and vigorously stirred for 10 minutes. The mixture was allowed to reach ambient temperature and stirred for 3 hours. The mixture was concentrated and the residue purified by silica gel chromatography (0-10% MeOH-DCM) to provide (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-(hydroxymethyl)benzyl)amino)isoindoline-1,3-dione as a yellow solid (4.23 g, 56%). LCMS (ESI) m/z 411.8 [M+H] +.
(S)-4-((4-(Chloromethyl)-2-fluorobenzyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione: A solution of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-(hydroxymethyl)benzyl)amino)isoindoline-1,3-dione (0.727 g, 1.77 mmol) in dry NMP (6 mL) was cooled to 0° C. and methane sulfonyl chloride (0.275 mL, 3.35 mmol) and DIEA (0.617 mL, 3.53 mmol) were added sequentially. The reaction mixture was allowed to reach ambient temperature and was stirred for 18 hours. The reaction mixture was slowly added to H 2O (60 mL) cooled to 0° C. with vigorous mixing. The resulting suspension was filtered and the collected solid was washed with H 2O and Et 2O. The solid was dissolved in EtOAc and the solution dried with MgSO 4, filtered and concentrated to provide (S)-4-((4-(chloromethyl)-2-fluorobenzyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione as a yellow solid (0.600 g, 79%). LCMS (ESI) m/z 430.0 [M+H] +.
(S)-2-(2,6-Dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dione: To a solution of (S)-4-((4-(chloromethyl)-2-fluorobenzyl)amino)-2-(2,6-dioxopiperidin-3-yl)isoindoline-1,3-dione (300 mg, 0.698 mmol) in dry DMSO (1.0 mL) was added 4-(azetidin-3-yl)morpholine hydrochloride (125 mg, 0.698 mmol) and DIEA (0.122 mL, 0.698 mmol). The reaction mixture was stirred at ambient temperature for 18 hours and was diluted with DMSO (1 mL). The solution was purified by chiral reverse-phase chromatography to give (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline-1,3-dione (89 mg, 24%, 97% ee). LCMS (ESI) m/z 536.2 [M+H] +.
Example 1: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione
[00242] Synthesis of Ethyl 4-nitro-1,3-dioxo-isoindoline-2-carboxylate (Compound 10): To a solution of 4-nitroisoindoline-1,3-dione (Compound 11, 440 g, 2.29 mol) and TEA (262 g, 2.59 mol, 359 mL) in dry DMF (2.2 L) was cooled to 0 °C and ethyl chloroformate (313 g, 2.89 mol, 275 mL) was added dropwise over 5 minutes. The reaction mixture was stirred at 22 °C for 10 hours. The mixture was slowly added to chilled water (10 L) and the resulting suspension stirred for 5 minutes. The suspension was filtered and the filter cake was washed with water (1 L). The solid was dissolved with ethyl acetate (5 L) and the organic phase was washed with aqueous HCl (1 M, 1 L), water (2 L) and brine (2 L). The organic phase was dried over sodium sulfate , filtered and concentrated to give Compound 10 (360 g, 59%) as a white solid. 1 H NMR (400 MHz CDCl 3 ) δ ppm 8.24 (d, J = 7.6 Hz, 1H), 8.19 (d, J = 8.4 Hz, 1H), 8.06-8.02 (m, 1H), 4.49 (q, J = 7.2 Hz, 2H), 1.44 (t, J = 6.8 Hz, 3H).
[00243] Synthesis of tert-Butyl (4S)-5-amino-4-(4-nitro-1,3-dioxo-isoindolin-2-yl)-5-oxo-pentanoate (Compound 6): To a solution of Compound 10 (165 g, 625 mmol) and DIEA (113 g, 874 mmol, 153 mL) in dry THF (1700 mL) was added tert-butyl (4S)-4,5-diamino-5-oxo-pentanoate hydrochloride ( 149 g, 625 mmol) and heated at reflux for 10 hours. The reaction mixture was concentrated under reduced pressure. The resulting residue was diluted with methyl tert-butyl ether (5 L) and stirred at 20 °C for 1 hour. The suspension was filtered and the filter cake was dissolved with DCM (4 L). The organic phase was washed with water (1.5 L x 3), brine (1.5 L) and dried over sodium sulfate . The organic phase was filtered and concentrated under reduced pressure to give a light yellow oil. The oil was diluted with hexane / ethyl acetate (10/1, 2 L) and stirred until a light yellow suspension formed. The suspension was filtered and the filter cake was triturated and concentrated in vacuum to give Compound 6 (175 g, 74%) as a light yellow solid. 1 H NMR (400 MHz CDCl3) δ ppm 8.12 (d, J = 8.0 Hz, 2H), 7.94 (t, J = 8.0 Hz, 1H), 6.48 (s, 1H), 5.99 (s, 1H), 4.84- 4.80 (m, 1H), 2.49-2.44 (m, 2H), 2.32-2.27 (m, 2H), 1.38 (s, 9H).
[00244] Synthesis of tert-Butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): To a suspension of Compound 6 (170.0 g, 450.5 mmol, 1.00 eq) in DMA (1.00 L) was added palladium on carbon (50.0 g, 10% purity) under nitrogen. The suspension was degassed under vacuum and purged with hydrogen gas several times. The mixture was stirred under hydrogen gas (50 psi) at 25 °C for 16 hours. The mixture was filtered and the filtrate was poured into cooled water (3.0 L). The mixture was stirred at 10 °C for 1 hour and filtered. The filter cake was washed with water (700 mL) and dissolved in DCM (1.00 L). The organic phase was dried over sodium sulfate , filtered and concentrated under reduced pressure to give Compound 5 (107 g, 68%) as a green solid. 1 H NMR (400 MHz DMSO-d 6 ) δ ppm 7.52 (s, 1H), 7.43 (dd, J = 8.4, 7.2 Hz, 1H), 7.13 (s, 1H), 6.95-6.99 (m, 2H), 6.42 (s, 2H), 5.75 (s, 1H), 4.47-4.51 (m, 1H), 2.32-2.33 (m, 1H), 2.14-2.20 (m, 3H), 1.32 (s, 9H); HPLC purity, 100.0%; SFC purity, 100.0% ee.
[00245] Synthesis of 2-fluoro-4-(hydroxymethyl)benzaldehyde (Compound 8): To a solution of 4-(((tert-butyldimethylsilyl)oxy)methyl)-2-fluorobenzaldehyde (370.0 g, 1.38 mol, 1.00 eq ) in THF (1.85 L) was added a solution of p-toluenesulfonic acid monohydrate (78.7 g, 413.6 mmol, 0.30 eq) in water (1.85 L) drop-wise at 10 °C. The mixture was stirred at 27 °C for 16 hours. TEA (80 mL) was added drop-wise and stirred for 10 minutes. The organic phase was separated and the aqueous phase was extracted with ethyl acetate (600 mL × 4). The combined
organic phase was washed with brine (1.50 L), dried over anhydrous sodium sulfate , filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 8 (137.5 g, 76%) as a yellow oil. 1 H NMR (400 MHz CDCl 3 ) δ ppm 10.34 (s, 1H), 7.86 (dd, J = 8.0, 7.2 Hz, 1H), 7.25 (s, 1H), 7.22 (d, J = 4.4 Hz, 1H) , 4.79 (d, J = 6.0 Hz, 2H), 1.91 (t, J = 6.0 Hz, 1H).
[00246] Synthesis of tert-Butyl (S)-5-amino-4-(4-((2-fluoro-4-(hydroxymethyl)benzyl)amino)-1,3-dioxoisoindolin-2-yl)-5- oxopentanoate (Compound 2-b): To a solution of Compound 5 (100.0 g, 287.9 mmol, 1.00 eq) and Compound 8 (57.7 g, 374.3 mmol, 1.30 eq) in dry DCM (1.00 L) was added TFA (164.1 g , 1.44 mol, 5.00 eq) at 0 °C. The reaction mixture was stirred at 28 °C for 2 hours. To the solution was added sodium cyanoborohydride (27.1 g, 431.8 mmol, 1.50 eq) at 0 °C. The mixture was stirred at 28 °C for 30 minutes. The reaction mixture was quenched by addition of MeOH (600 mL) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2-b (110.0 g, 74.0%) as a yellow solid. 1 H NMR (400 MHz, DMSO-d6) δ ppm 7.56 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.34 (t, J = 8.0 Hz, 1H), 7.02-7.18 ( m, 4H), 6.94-7.01 (m, 2H), 4.57 (d, J = 6.0 Hz, 2H), 4.47-4.53 (m, 3H), 2.31-2.35 (m, 1H), 2.15-2.22 (m, 3H), 1.31 (s, 9H); HPLC purity, 94.0%; SFC purity, 100.0% ee.
[00247] Synthesis of tert-butyl (S)-5-amino-4-(4-((4-(chloromethyl)-2-fluorobenzyl)amino)-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2-a): To a solution of Compound 2-b (100.0 g, 206.0 mmol, 1.00 eq) in NMP (430.0 mL) was added DIEA (79.9 g, 617.9 mmol, 3.00 eq) and MsCl (47.2 g, 411.9 mmol, 2.00 eq) at 0 °C. The ice bath was removed, and the reaction was stirred at 28°C for 10 hours. The reaction was poured into cooled water (<10°C, 2.0 L) and stirred for 10 minutes. The mixture was extracted with methyl tert-butyl ether (750 mL x 3). The combined organic layer was washed with brine (1.25 L), dried over sodium sulfate , filtered and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2-a (86.0 g, 81.2%) as a yellow solid.
[00248] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2): To a solution of 4-(azetidin-3-yl)morpholine hydrochloride (Compound 7 HCl, 30.5 g, 170.7 mmol, 1.00 eq) and DIEA (66.2 g, 512.0 mmol, 3.00 eq) in DMSO (350.0 mL) was added to a solution of Compound 2-a (86 g, 170.65 mmol, 1.00 eq) in DMSO (350.0 mL) drop-wise at 15 °C. The reaction mixture was stirred at 28 °C for 16 hours. The reaction mixture was poured into cold half saturated brine (<10°C, 2.5 L) and extracted with ethyl acetate (1.50 L, 1.00 L, 800.0 mL). The combined organic phase was washed with saturated brine (1.50 L), dried over sodium sulfate , filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to give Compound 2 (68.3 g, 65.7%) as a yellow solid. 1 H NMR (400 MHz DMSO-d 6 ) δ ppm 7.55 (s, 1H), 7.50 (dd, J = 8.4, 7.2 Hz, 1H), 7.31 (t, J = 8.0 Hz, 1H), 7.16 (s, 1H), 6.94-7.10 (m, 5H), 4.56 (d, J = 6.4 Hz, 2H), 4.49-4.52 (m, 1H), 3.54-3.55 (m, 6H) 3.31-3.32 (m, 3H), 2.81-2.88 (m, 3H), 2.29-2.38 (m, 1H), 2.15-2.25 (m, 7H), 1.30 (s, 9H); HPLC purity, 100.0%; SFC purity, 100.0% ee.
[00249] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione (Compound 1): A solution of Compound 2 (30.0 g, 49.2 mmol, 1.00 eq) and benzenesulfonic acid (31.1 g, 196.8 mmol, 4.00 eq) in acetonitrile (480.0 mL) was stirred at reflux for 3 hours. The reaction was cooled to 20 °C, poured into cold brine:saturated sodium bicarbonate solution (1:1, <10 °C, 2.0 L) and extracted with ethyl acetate (1.0 L). The organic phase was washed with cold brine:saturated sodium bicarbonate solution (1:1, <10°C, 1.00 L) once more. The combined aqueous phase was extracted with ethyl acetate (500.0 mL x 2). The combined organic phase was washed with cold brine (<10°C, 1.0 L), dried over sodium sulfate , filtered and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to give Compound 1 (17.5 g, 66.0%) as a yellow solid. 1 H NMR (400 MHz, DMSO-d 6 ) δ ppm 11.10 (s, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.04-7.10 (m , 4H), 7.00 (d, J = 8.4 Hz, 1H), 5.07 (dd, J = 12.8, 5.2 Hz, 1H), 4.58 (d, J = 6.4 Hz, 2H), 3.53-3.55 (m, 6H) , 3.30-3.32 (m, 2H), 2.81-2.89 (m, 4H), 2.54-2.61 (m, 2H), 2.20 (m, 4H) 2.03-2.06 (m, 1H); HPLC purity, 100.0%; SFC purity, 97.2% ee; LCMS (ESI) m/z 536.1 [M+H] + .
Example 2: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione
[00250] Synthesis of tert-butyl (S)-5-amino-4-(4-nitro-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 6): Ethyl acetate (245 mL, 5 V ), 3-nitrophthalic anhydride (49.1 g, 0.25 mol, 1 eq), and tert-butyl (S)-4,5-diamino-5-oxopentanoate hydrochloride (59.2 g, 0.25 mol,
1 eq) were charged into a reactor and cooled to 15-20°C. A premade solution of CDI (66.7 g,
0.41 mol, 1.5 eq) in DMF (245 mL, 5 V) was charged and the mixture was stirred at 20-25°C for
1 hour. The reaction was quenched with 15% (wt/wt) aqueous citric acid solution (10 V).
EtOAc (5 V) was added, the mixture was agitated and the phases split and separated. Tea
aqueous layer was extracted with EtOAc (5 V) and the combined organic layers were washed
twice with a 5% (wt/wt) aqueous citric acid solution (5 V each wash). The organic layer was
distilled at reduced pressure to 5 V and further continuously distilled at reduced pressure with the addition of iPrOH (10 V), maintaining a constant volume at 5 V. The final distillate was diluted to 13 V with iPrOH and used in the next step without further handling. 91% solution yield. [00251] Synthesis of tert-butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): The solution of Compound 6 in iPrOH was charged to a hydrogenation reactor. 10% palladium on carbon (50% wet, 4.65g 5 wt%) was charged. The reaction mixture was stirred under 50-60 psi H2 at 40-50 o C for 16 hrs. The reaction mixture was filtered and the filter cake was washed three times with iPrOH (1 V each wash). The solution was distilled at reduced pressure to 5 V, cooled to ambient temperature and seeded (1 wt%). Water (20 V) was charged at 20-25 o C. The resulting slurry was cooled to 3-8 o C for 4-8 hrs. The solids were collected by filtration and washed three times with cold water (1.5 V each wash). The solids were dried at 35-45 o C under reduced pressure to give Compound 5 in 87% yield. 1 H NMR (500 MHz DMSO-d 6 ) δ (ppm): 7.52 (s, 1H), 7.43 (dd, J = 8.4, 7.0 Hz, 1H), 7.13 (s, 1H), 6.97 (ddd, J = 10.9, 7.7, 0.61 Hz, 2H), 6.43 (s, 2H), 4.49 (m, 1H), 2.33 (m, 1H), 2.17 (m, 3H), 1.32 (s, 9H); HPLC purity, 99.2%; Chiral purity, 99.9% ee; LCMS (ESI) m/z 348.2, [M+H] + , 292.2 [Mt-Bu+H] + . Residual IPA: 0.7 mol% by 1 H NMR.
[00252] Synthesis of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine (Compound 4): A mixture of 4-bromo-3-flurobenzaldehyde (Compound 14, 82 g, 396 mmol ) and 4-(azetidin-3-yl)-morpholine hydrochloride (Compound 7 HCl, 72 g, 396 mmol) in acetonitrile (820 ml) was agitated at 25±5°C for at least 3 hours. The mixture was cooled to 10±5°C and sodium triacetoxyborohydride (130 g, 594 mmol) was added in four portions while maintaining the temperature of the mixture below 30°C. The temperature of the mixture was adjusted to 25±5°C and stirred for at least 30 min until reaction completion. The mixture was transferred to a precooled (10-15°C) solution of aqueous citric acid (152 g in 400 ml water, 792 mmol) while maintaining the temperature below 30°C. Once the quenching process was complete, the mixture was concentrated to ~ 560 ml (7 volumes) while keeping the temperature at or below 45°C. The mixture was then washed with toluene (320 ml). To the aqueous phase was added THF and the pH was adjusted to above 12 with aqueous NaOH solution (320 ml, 10 N). The phases were separated, and the aqueous phase was removed. The organic phase was washed with brine and subsequently concentrated with addition of THF (~ 3L) until KF ≤ 0.10%. The mixture was filtered to remove any inorganics and the product Compound 4 was isolated as a solution in THF with 95% yield.
[00253] Synthesis of sodium (2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)phenyl) (hydroxy)methanesulfonate (Compound 13): A solution of Compound 4 (520 g, 1.58 mol) in
THF (380 ml) was cooled to −15 ± 5 °C. A solution of iPrMgCl . LiCl (1.3 M, 1823 ml, 2.37 mol) in THF was added over the course of at least 1 hour while maintaining the temperature below −10 °C. After addition was complete, the temperature of the reaction mixture was adjusted to 0 ± 5 °C and stirred for at least 1 hour. Once magnesiation was complete, the mixture was cooled to −15 ± 5 °C (target −15 °C to −20 °C) and a solution of DMF (245 ml g, 3.16 mol) in THF (260 ml) was added slowly over the course of at least 1 hour while maintaining the temperature below −10 °C. The temperature of the mixture was then adjusted to −15 ± 5 °C and agitated for at least 4 hours.
[00254] Upon reaction completion, the reaction mixture was charged into an aqueous 3 N HCl solution (2600 ml) over the course of at least 1 hour while maintaining the temperature below −5 °C. The temperature of the mixture was then adjusted to 5 ± 5 °C and agitation was stopped, letting the mixture settle for at least 15 minutes. The layers were separated. The lower aqueous layer containing the product was washed with 2-MeTHF (2600 ml). The aqueous layer was then charged with 2-MeTHF (2600 ml) and the temperature of the batch was adjusted to −10 ± 5 °C. To the cooled mixture, an aqueous 5 N NaOH (728 ml, 3.64 mol) solution was added while maintaining the temperature below −5 °C until the pH of the mixture was between 10 and 11. The temperature of the mixture was adjusted to 5 ± 5 °C and agitated for at least 15 minutes. The agitation of the mixture was stopped and the mixture allowed to settle for at least 15 minutes. The layers were separated, and the lower aqueous layer was back extracted two times with 2-MeTHF (2600 ml). The combined organic layer was washed with water (1040 mL) and the organic solution was evaporated to dryness, affording 372 g of crude Compound 3 freebase as an oil (yield 85%). 1 H NMR (DMSO-d 6 ) δ (ppm): 10.18 (s, 1H), 7.78 (t, J =7.7 Hz, 1H), 7.23-7.35 (m, 2H), 3.66 (s, 2H), 3.51 -3.60 (m, 4H), 3.26-3.47 (m, 2H), 2.72-2.97 (m, 3H), 2.12-2.32 (m, 4H).
[00255] The crude Compound 3 freebase (4.3 kg) was adsorbed onto silica gel (8.6 kg) with 100% DCM, loaded onto a 60 L column containing 12.9 kg silica gel (packed with 100% DCM), and eluted with DCM ( 86 L), followed successively by 1% MeOH/DCM (40 L), 3% MeOH/DCM (80 L) and 10% MeOH/DCM (40 L). The fractions were collected and concentrated at or below 38˚C to give Compound 3 as a purified oil (3.345 kg, yield 66%).
[00256] A portion of Compound 3 (1.0 kg, 3.59 mol) was dissolved in ethanol (16.0 L, 16
vol) at 20±5 °C and the mixture heated to 40 °C. A solution of Na2S2O5 (622.0 g, 3.27 mol; 0.91 eq) in water (2 L, 2 vol) was prepared at 20±5 °C and added to the freebase solution at 40 °C to obtain an off-white suspension. The batch was agitated and maintained at 40 °C for 2 hrs, then cooled to 20±5 °C and agitated for 1 to 2 hrs. The batch was filtered and washed with ethanol (2×2.0 L, 2×2 vol) to obtain an off-white solid. The wet cake was dried under vacuum at 40 °C for 18 hrs to afford about 1.88 kg of Compound 13.
[00257] Synthesis of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde (Compound 3): Compound 13 (1.88 kg) was dissolved in ethyl acetate (15.0 L) at 20±5 °C . A 2 M Na 2 CO 3 solution (total 15.0 L used) was added to adjust the pH to 10.0. The batch was agitated for 1 to 1.5 hrs at 20±5 °C. After the reaction was complete, the phases were separated and the organic layer was washed with brine (2.0 L). The organic layer was concentrated to dryness at 35-38 °C to afford 852.0 g of Compound 3 as a colorless oil (yield 81%).
[00258] Synthesis of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde bis-oxalic acid salt (Compound 3 bis-oxalic acid salt): A portion of the Compound 3 oil (187 g, 0.67 mol) was dissolved in isopropanol (1125 ml) and water (375 ml). A first portion (~30%) of this freebase mixture (480 ml) was slowly added over the course of at least 30 minutes to a solution of oxalic acid (125 g, 1.38 mol) in IPA (1125 ml)/water (375 ml) at 60 ± 5 °C. A second portion (~20%) of the freebase mixture (320 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C. The reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. A third portion (~25%) of the freebase mixture (~ 400 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C and the reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. The remaining freebase solution (400 ml) was slowly added over the course of at least 30 minutes to the reaction mixture at 60 ± 5 °C and the reaction mixture was agitated at 60 ± 5 °C for at least 90 minutes. The temperature of the mixture was adjusted to 20 ± 5 °C (target 20 °C) over the course of at least 1 hour and the mixture was agitated for at least 16 hours at 20 ± 5 °C and then filtered. The cake was washed three times with IPA (2 x 375 ml) and dried in the drying oven at ≤ 40 °C with a slow bleed of nitrogen to afford 261 g of Compound 3 bis-oxalic acid salt (yield 85%). 1 H NMR (DMSO-d 6 ) δ (ppm): 10.21 (s, 1H), 7.87 (t, J = 7.6 Hz, 1H), 7.42-7.56 (m, 2H), 4.31 (s, 2H), 3.89 -4.03 (m, 2H), 3.75-3.89 (m, 2H), 3.60 (br t, J = 4.3 Hz, 4H), 3.26 (br t, J = 6.9 Hz, 1H), 2.37 (br s, 4H) .
[00259] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2):
Acetonitrile (6.8 L, 8.0 X Vol) was added to a 30 L jacketed cylindrical reactor. Compound 5 (0.845 kg, 1.00 X Wt) and Compound 3 bis-oxalic acid salt (1.35 kg, 1.60 The contents of the reactor were equilibrated with agitation at 20 ± 5 °C. Trifluoroacetic acid (0.19 L, 0.22 X Vol) was added dropwise, maintaining the batch temperature at 20 ± 5 °C. The reaction mixture was stirred at 20 ± 5 °C for no less than 5 minutes and then sodium triacetoxyborohydride (0.13 kg, 015 X Wt) was added as a solid, maintaining the batch temperature at 20 ± 5 °C. The process of adding trifluoroacetic acid and then sodium triacetoxyborohydride was repeated an additional 5 times. After the last addition, the reaction mixture was sampled to determine the reaction progress. The reaction was held at 20 ± 5 °C overnight. The reaction mixture was then quenched with water (3.4 L, 4.0 X Vol), maintaining the batch temperature at 20 ± 5 °C. The mixture was then stirred at 20 ± 5 °C for no less than 30 minutes and the resulting slurry filtered through a 3 L sintered glass filter, directing the filtrates to clean containers. The reactor was rinsed with acetonitrile (0.4 L, 0.5 X Vol) and the rinse passed through the contents of the 3 L sintered glass filter, directing the filtrate to the containers containing the main batch. The contents of the containers were concentrated to ~5 X Vol under reduced pressure at a bath temperature of no more than 30 °C. The residue was transferred to a clean reactor, was rinsing with 2-MeTHF (2.5 L, 3.0 X Vol) to complete the transfer. Additional 2-MeTHF (10.1 L, 12.0 X Vol) was added to the reactor, followed by water (3.4 L, 4.0 X Vol). The mixture was agitated for no less than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes at 20 ± 5 °C before transferring the bottom aqueous layer to new containers. An aqueous sodium bicarbonate solution (5.3 L, 6.3 X Vol, 9% wt/wt) was added to the reactor with stirring over 30 minutes, maintaining batch temperature no more than 25 °C. The mixture was agitated for no more than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes at 20 ± 5 °C before the bottom aqueous layer was transferred to new containers. The aqueous sodium bicarbonate wash was repeated an additional 2 times to reach a pH of about 6.6 for the spent aqueous layer. A saturated aqueous solution of NaCl(0.85 L, 1.0 X Vol) was then added to reactor with agitation. The mixture was agitated for no less than 15 minutes at 20 ± 5 °C, then allowed to settle for no less than 10 minutes before the bottom aqueous layer was transferred to new containers. The remaining organics were concentrated under reduced pressure to a batch volume of ~5 X Vol at a bath temperature of about 40 °C. Acetonitrile (5.1 L, 6.0 X Vol) was added to the residual volume and the resulting solution concentrated to a batch volume of ~ 5 X Vol under reduced pressure at bath temperature of about 40 °C. The process of adding acetonitrile and concentrating under vacuum was repeated two more times to reach the distillation endpoint with a water content of about 1%. The acetonitrile solution was transferred to a clean container along with two 1.7 L (2.0 X Vol) rinses and held at 5 °C overnight. The acetonitrile solution was then filtered through a 3 L sintered glass filter, followed by a 1.7 L (2.0 X Vol) acetonitrile rinse, directing the filtrates to a clean container. The filtrate was transferred to a clean reactor and the container rinsed twice with 1.7 L (2.0 X Vol) of acetonitrile to complete the transfer. Enough acetonitrile (roughly 0.6 L) was added to adjust the total volume in the reactor to about 14 L. A solution assay of the contents of the reactor was obtained to calculate the amount of Compound 2 present for use in the next step (result = 1.3 kg = 1.00 X Wt for remainder of process).
[00260] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione bis-besylate salt (Compound 1 bis-besylate salt): The solution of Compound 2 in acetonitrile from the previous step was diluted with acetonitrile (roughly 2 L) such that the total volume in the reactor was about 16 L. The solution was cooled with stirring to 10 ± 5 °C and held within that range for 96 hours. Benzenesulfonic acid (1.86 kg, 1.43 X Wt) was added while sparging the reaction mixture with nitrogen gas and maintaining the batch temperature at 10 ± 10 °C. The temperature of the reactor was then adjusted to 20 ± 5 °C and the mixture stirred at that temperature for 60 minutes. The total volume of reaction mixture was adjusted back to 16 L to account for solvent lost during sparging by the addition of acetonitrile (roughly 0.4 L). The reaction mixture was then heated to 55 ± 5 °C over the course of about 30 minutes and held in that range for 15 to 16 hours for reaction completion. The mixture was then cooled to 50 ± 5 °C and MTBE (3.9 L, 3.0 X Vol) was added, maintaining the batch temperature at 50 ± 5 °C. The mixture was allowed to stir at 50 ± 5 °C for about 1.5 hours to establish a self-seeded slurry. Additional MTBE (3.9 L, 3.0 X Vol) was added to the reactor over the course of about 1.75 hours at 50 ± 5 °C. The slurry was cooled to 20 ± 5 °C over the course of about 1.75 hours and held in that temperature range overnight. The slurry was filtered using a Buchner funnel. The reactor was rinsed twice with
MTBE (3.9 L each, 3.0 X Vol) and the rinse was used to wash the solids in the Buchner funnel. The solids were dried on drying trays for about 23 hours at 40 °C under reduced pressure (15-150 mbar), yielding 1.62 kg (77.9%) of Compound 1 bis-besylate salt.
[00261] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione hydrochloride salt (Compound 1 HCl): A suspension of Compound 1 bis-besylate salt (120 g, 1 equiv.) in 2-MeTHF (25 L/kg) was added to a reactor and agitated at 10 °C. A solution of KHCO 3 (32.5 g, 2.4 equiv) in water (1.8 L, 6 L/kg) was added to the slurry over the course of 40 minutes. The mixture was stirred for an additional 30 minutes. The batch was then allowed to settle, at which point the aqueous (bottom) layer was separated and discarded. An aqueous solution of NaCl (5%, 5 L/kg, 575 ml) was added to the organic layer and the mixture was agitated for 10 minutes, after which point the temperature was raised to 20 °C. The batch was allowed to settle, at which point the aqueous (bottom) layer was discarded. The brine was repeated a second time.
Additional 2-MeTHF (500 ml) was added to dilute the organic layer, resulting in a concentration of about 20 mg product per ml. A solution of HCl (total 0.98 eq.) in 2-MeTHF was prepared and a portion (20% of total, corresponding to ~0.2 eq.) then added to the reaction mixture over the course of about 10 min. Seeds of Compound 1 hydrochloride (~5% wt) were added, but did not dissolve. The batch was held under vigorous agitation for one hour. To the slurry, the remaining portion of the HCl solution (~0.78 eq.) was added over the course of 3 hours at a constant rate. Vigorous agitation was maintained. After addition was complete, the batch was held for one hour, after which the batch was filtered, washed three times with 3 L/kg of 2-MeTHF . The filter cake was placed in a vacuum oven at 22 °C for 12 hours, at which point the temperature was raised to 40 °C. Dry cake of Compound 1 hydrochloride (58g, 75% yield) was obtained and packaged. Achiral HPLC purity: 98.91%; chiral HPLC purity: 99.68%.
Example 4: Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3- morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione
[00264] Synthesis of tert-butyl (S)-5-amino-4-(4-nitro-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 6): To a solution of 3-nitrophthalic anhydride (Compound 12,
mL) and 2-MeTHF (110 mL) at 25°C. 2,6-Lutidine (23.4 mL, 201 mmol, 1.14 eq) was added
slowly to maintain the temperature at or below 25°C. The mixture was aged at 25°C for 1 hour before being cooled to 5°C. CDI (4.17 g, 25.7 mmol, 0.146 eq) was added and stirred until the temperature returned to 5°C. Another portion of CDI (4.62 g, 28.5 mmol, 0.161 eq) was added and stirred until temperature returned to 5°C. CDI (8.87 g, 54.7 mmol, 0.310 eq) was added and stirred until the temperature returned to 5°C. CDI (8.91 g, 54.9 mmol, 0.311 eq) was added and stirred until the temperature returned to 5°C. The mixture was warmed to 20°C and CDI (16.4 g, 101.1 mmol, 0.573 eq) was added, and the mixture was aged at 20°C for 16 hours. The mixture was cooled to 5°C and a solution of 30 wt% citric acid and 5 wt% NaCl (350 mL) was added slowly while maintaining the temperature. The mixture was warmed to 20°C and aged for 30 minutes. The phases were split and separated. The organic phase was diluted with EtOAc (175 mL) and washed with a solution of 5 wt% citric acid (175 mL), and concentrated by distillation (75 torr, 50°C) to a volume of 175 mL EtOAc. The solvent was changed to iPrOH by constant volume distillation (75 torr, 50°C) with 350 mL iPrOH to a final volume of 175 mL. The distillate was diluted with 200 mL iPrOH to afford Compound 6 as a solution for use in the next step. 1 H NMR (500 MHz, CDCl 3 ) δ (ppm): 8.18 – 8.13 (m, 2H), 7.96 (t, J = 7.8 Hz, 1H), 6.34 (s, 1H), 5.59 (s, 1H), 4.90 (dd, J = 10.1, 4.6 Hz, 1H), 2.61 (ddt, J = 14.6, 10.1, 6.1 Hz, 1H), 2.49 (ddt, J = 14.2, 8.7, 5.2 Hz, 1H), 2.44 – 2.29 ( m, 2H), 1.44 (s, 9H).
[00265] Synthesis of tert-butyl (S)-5-amino-4-(4-amino-1,3-dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 5): To a solution of Compound 6 in iPrOH (375 mL) was added 5% palladium on carbon (1.23 g, 3.5 wt%, wet). The mixture was purged with nitrogen five times and with hydrogen three times. The mixture was pressurized with hydrogen (50 psi) and aged at 50°C for 16 hours. The mixture was cooled to room temperature and purged with nitrogen three times, filtered to remove catalyst, and the filter cake was washed with iPrOH (20mL) three times. The filtrate was concentrated to 200 mL, seeded (0.454 g, 1.3 wt%) at 22 °C, and aged for 45 minutes. Water (1325 mL) was added over 3 hours at 22°C. After the addition of water, the mixture was cooled to 8°C over 2 hours and aged for 1 hour at 8°C. The slurry was filtered, and the cake was rinsed with cold water (200 mL) three times and dried under vacuum at 50°C to yield Compound 5 as a yellow solid (47.97 g, 80.6% yield, 99.62% LC purity, 103% 1 H NMR potency). 1 H NMR (500 MHz, CDCl 3 ) δ (ppm): 7.46 (dd, J = 8.3, 7.0 Hz, 1H), 7.19 (d, J = 7.2 Hz, 1H), 6.89 (d, J = 8.3 Hz, 1H), 6.28 (s, 1H), 5.41 (s, 1H), 5.28 (s, 2H), 4.83 (dd, J = 9.3, 6.0 Hz, 1H), 2.52 (p, J = 7.0 Hz, 2H), 2.36 – 2.29 (m, 2H), 1.44 (s, 9H). 13 C NMR (126 MHz, CDCl 3 ) δ (ppm): 171.80, 171.12, 169.64, 168.27, 145.70, 135.50, 132.20, 121.43, 112.98, 80.99, 53.04, 32.23, 28.02 , 24.36. LCMS (ESI): m/z 291.9 [M+H – tBu]
[00266] Synthesis of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine bis-methanesulfonic acid salt (Compound 4 bis-methanesulfonic acid salt): A mixture of 4-bromo-3- fluorobenzaldehyde (Compound 14, 102 g, 493 mmol) and 4-(azetidin-3-yl)morpholine hydrochloride (Compound 7 HCl, 90 g, 493 mmol) in acetonitrile (1000 ml) was agitated at a temperature of about 20 to 25 °C for 2 to 3 hours. The slurry was cooled to temperature of about 10 to 15 °C and sodium triacetoxyborohydride (STAB, 162 g, 739 mmol) was added in 4 portions over the course of about 45 minutes while maintaining the batch temperature at no more than 30 °C. The slurry was stirred at a temperature of about 20 to 25 °C for at least 30 minutes and then quenched by an aqueous citric acid solution (191 g, 986 mmol in 500 ml of water) at a temperature of about 40 to 45 °C over the course of 2 hours. Upon completion of the quenching process, the batch volume was reduced by vacuum distillation to about 700 ml at a temperature of no more than 45 °C. Cyclopentylmethylether (CPME, 400 ml) was added to the aqueous solution to afford a final volume of about 1100 ml. The pH was adjusted to about 8 to 9 by addition of an aqueous solution of 10 N NaOH (added volume about 430 ml). The phases were separated, and the aqueous phase discarded. The organic phase was washed with brine (100 ml) twice such that the pH was no more than 8 and the volume was adjusted to about 1000 ml with addition of extra CPME. The batch was distilled at constant volume under reduced pressure with addition of CPME until KF was no more than 0.15%. CPME was added (if needed) to adjust the batch to a volume of 1000 ml at the end of distillation. The dry CPME solution was seeded (500 to 750 mg) at ambient temperature. The seeded, dry CPME slurry was heated to a temperature of 50 to 60 °C and then charged with methanesulfonic acid in 200 ml of CPME over the course of 4 to 5 hours. The slurry was then cooled to 20 °C over the course of 4 to 5 hours and kept at 20 °C for 3 to 4 hours, filtered, rinsed with CPME and dried in a vacuum oven at 35 to 40 °C over 16 hours to give Compound 4 bis-methanesulfonic acid salt as a white solid. 1 H NMR (500 MHz DMSO-d 6 ) δ (ppm): 10.62 (br s, 1-2H), 7.85 (t, J = 7.8 Hz, 1H), 7.58 (dd, J = 9.5 Hz, 1.9 Hz, 1H), 7.34 (dd, J = 8.2 Hz, 1.8 Hz, 1H), 4.55-4.24 (m, 7H), 3.84 (br s, 4H), 3.14 (m, 4H); HPLC purity, 99.8%, LCMS (ESI) m/z 329.1 /331.1[M/M+2] + . XRPD pattern of the product is shown in FIG.10. DSC thermogram of the product is shown in FIG.11.
[00267] Preparation of 4-(1-(4-bromo-3-fluorobenzyl)azetidin-3-yl)morpholine (Compound 4): A slurry of Compound 4 bis-methanesulfonic acid salt (70 g, 134 mmol) in t -butyl methyl ether was cooled to 10±5°C. An aqueous solution of NaOH (2 N, 201 ml, 403 mmol) was added over the course of at least 30 minutes while maintaining the batch temperature at about 15 °C. After the addition of NaOH , the batch temperature was raised to 20±5°C and agitated over the course of about 20 minutes. The organic layer was separated and washed with water (210 ml) three times. The organic layer was subsequently concentrated with addition of THF (~ 1.05L) until KF ≤ 0.10%. The product Compound 4 was isolated as a solution in THF
with 95% solution yield.
[00268] Preparation of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde dihydrochloride (Compound 3 di-HCl): A solution of Compound 4 (44 g, 134 mmol) in THF (total volume ~ 350 ml) was then cooled to −20 ± 5 °C. A solution of iPrMgCl . LiCl (1.3 M, 176 ml, 228 mmol) in THF was added over the course of half an hour while maintaining the temperature below −10 °C. After the addition was complete, the batch was stirred at −20 ± 5 °C for 16 to 22 hours. DMF (21 ml, 268 mmol) was then added slowly over the course of 30 minutes while maintaining the batch temperature no more than -15 °C. The batch was stirred at −20 ± 5 °C for 6 to 24 hours. 2-MeTHF (350 ml) was then added to the batch over the course of 30 minutes, followed by slow addition of 3 N HCl (235 ml, 704 mmol) while keeping the batch temperature no more than -10 °C. After the addition of aqueous HCl, the batch was warmed to 0 ± 5 °C and 2 N aqueous NaOH (154 ml, 309 mmol) was added slowly to adjust the solution pH to about 8 to 9. The batch was stirred for about 30 minutes and then warmed to 20 ± 5 °C. The organic layer was separated and washed with 15% aqueous NaCl (3 x 140 ml). The organic layer was subsequently concentrated with addition of 2-MeTHF until KF ≤ 0.10%.
[00269] A portion of the free base of 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde (37.4 g, 134 mmol) so obtained was dissolved in 2-MeTHF (total ~ 420 ml) , to which isopropanol (420 ml) and water (21 ml) were added at 20 ± 5 °C. The batch was then heated to 50 ± 5 °C and a solution of HCl in IPA (5 to 6 N, 28 ml, half of total HCl volume) was added over the course of 1 hour. The batch was seeded with 2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzaldehyde dihydrochloride (700 mg) and aged for 1 hour. The remaining HCl (28 ml) was then added over the course of 1 hour. The batch was agitated at 50 ± 5 °C for 4 hours and then cooled to 20 ± 5 °C for 8 hours. The slurry was filtered, washed with IPA (210 ml), and the filter cake dried under vacuum at 50 ± 5 °C to afford Compound 3 dihydrochloride salt (36 g, yield 75%). 1 H NMR (DMSO-d 6 ) δ (ppm): 12.32-12.55 (m, 1H), 10.23 (s, 1H), 7.93 (t, J =7.6 Hz, 1H), 7.66 (d, J = 10.5 Hz , 1H), 7.58 (d, J = 7.9 Hz, 1H), 4.80 (br s, 2H), 4.48-4.70 (m, 2H), 4.30 (br s, 4H), 3.78-4.00 (m, 5H), 2.93-3.15 (m, 2H). Two polymorphic forms were obtained. XRPD pattern and DSC thermogram of Form A (anhydrous) are shown in FIG.5 and FIG.6, respectively. XRPD pattern, TGA thermogram and DSC thermogram of Form B (hydrate) are shown in FIG.7, FIG.8, and FIG.9, respectively.
[00270] Synthesis of tert-butyl (S)-5-amino-4-(4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)-1,3- dioxoisoindolin-2-yl)-5-oxopentanoate (Compound 2): A mixture of Compound 5 (12 g, 34.5 mmol, 1.0 eq) and Compound 3 dihydrochloride (14.56 g, 41.5 mmol, 1.2 eq) in MeCN (96 ml) was cooled to 0-5 °C. Trifluoroacetic acid (TFA, 2.0 ml, 26 mmol, 0.75 eq) was added, followed by sodium triacetoxyborohydride (STAB, 2.75 g, 12.95 mmol, 0.375 eq) while maintaining the internal temperature below 10 °C. The addition of TFA and STAB was repeated three additional times. After a total of four additions of TFA and STAB, the reaction was aged at 0-5°C for 1 hour. A 10% brine solution (108 ml) was then added to the reaction mixture over the course of 1 hour and partitioned with IPAc (96 ml). The mixture was warmed to 20-25 °C and aged for 30 minutes. The layers were then separated and the organic layer was washed with 2.0 M K3PO4 (114 ml). The pH of the spent aqueous layer should have a pH of about 8.5 – 9.0. The layers were separated again and the organic phase was washed with 8.5% NaHCO 3 (2 x 60 ml), with 30 minutes between each wash, followed by 24% brine (60 ml). The organic fraction was distilled to 72 ml at an internal temperature near 50°C. Toluene (72 ml) was added to bring the volume to 144 ml and distillation continued at constant volume at 50°C with feed and bleed until water content < 0.1. The mixture was heated to 50°C and acetonitrile (48 ml) was added, followed by slow addition of heptane (144 ml) while maintaining the internal temperature above 45 °C. The reaction was held at 50 °C for 2 hours. Once complete, the reaction was slowly cooled to 20-25°C over the course of 4 hours and held at 20-25°C overnight (16 hour). The yellow slurry was then filtered and the yellow cake displacement washed with 1:3:3 mixture of acetonitrile/heptane/toluene (3 x 48 ml). The final cake was then dried under reduced pressure at 50 °C under nitrogen to provide Compound 2 (87.7% isolated molar yield) with >99.0% LCAP. HPLC purity, 99.85%; Chiral purity, >99.9% ee. 1 H NMR (DMSO-d6, 500 MHz) δ (ppm) 7.55 (s, 1H), 7.51 (dd, J = 7.2, 8.4 Hz, 1H), 7.32 (t, J = 7.9 Hz, 1H), 7.16 ( s, 1H), 7.0-7.1 (m, 5H), 4.57 (d, J = 6.3 Hz, 2H), 4.5-4.5 (m, 1H), 3.5-3.6 (m, 6H), 3.3-3.4 (m, 3H), 2.8-2.9 (m, 3H), 2.3-2.4 (m, 1H), 2.1-2.3 (m, 7H), 1.31 (s, 9H); LCMS m/z 610.3 [M+H] + .
[00271] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione bis-besylate salt (Compound 1 bis-besylate): To a suspension of Compound 2 (130 g, 1.0 equiv.) in MeCN (1.56 L, 12 L/kg) agitated at 55 °C was added a solution of benzene sulfonic acid (185 g, 5.5
equiv.) in MeCN (0.39 L, 3 L/kg) and water (0.01 L, 2.0 equiv.). The mixture was stirred at 55 °C for 16 hours. After the reaction age, crystalline seeds (1.3 g, 1 wt%) of bis-besylate salt of Compound 1 were charged into the batch, resulting in formation of a yellow slurry. The slurry was then cooled to 20 °C over the course of 90 minutes. 2-MeTHF (1.3 L, 10 L/kg) was added to the batch slowly over 2 hours at 20 °C. The batch was agitated for an additional 4 hours at 20°C. The yellow slurry was then filtered and the yellow cake re-slurried with MeTHF (1.3 L, 10 L/kg) followed by a displacement MeTHF (0.65 L, 5 L/kg) wash. The final cake was then dried under reduced pressure at 50 °C under nitrogen to give Compound 1 bis-besylate salt (160 g, 88.4% yield). HPLC purity: 98.39%; chiral HPLC purity: 100%. XRPD pattern, TGA thermogram and DSC thermogram of the product are shown in FIG.1, FIG.2, and FIG.3, respectively.
[00272] Synthesis of (S)-2-(2,6-dioxopiperidin-3-yl)-4-((2-fluoro-4-((3-morpholinoazetidin-1-yl)methyl)benzyl)amino)isoindoline -1,3-dione hydrochloride salt (Compound 1 HCl): A suspension of Compound 1 bis-besylate salt (300 g, 1 equiv.) in EtOAc (4.68 L, 15.6 L/kg) and 2-propanol (0.12 L, 0.4 L/kg) was agitated at 15 °C. To the suspension was added a solution of KHCO 3 (82.4 g, 2.5 equiv) in water (1.8 L, 6 L/kg) over 30 minutes. The mixture was heated to 20 °C over 30-60 minutes and then agitated for 30 minutes. The batch was allowed to settle for 30 minutes, at which point the aqueous (bottom) layer was discarded. To the rich organic layer was added water (1.2 L, 4 L/kg) and the reactor contents were agitated for 30 minutes. The batch was allowed to settle for 30 minutes, at which point the aqueous (bottom) layer was discarded. To the rich organic stream was added 2-propanol (2.375 L, 7.9 L/kg) and the stream was then filtered. Water was added to the filtrate to adjust the water content to 8≤KF≤8.2. To the above agitated solution at 20 °C was added 0.2 N HCl (38 mL, 0.025 equiv prepared in EtOAC/IPA 2:1, v/v with 8wt% water) over 10 minutes. To the mixture was added crystalline seeds of Compound 1 hydrochloride salt (1.6 g, 0.5wt%) and the contents of the reactor were agitated at 20 °C for 30 minutes. To the suspension was added 0.2 N HCl (1.44 L, 0.945 equiv. prepared in EtOAC/IPA 2:1, v/v with 8 wt% water) over 4.5 hours. The slurry was agitated for 14 hours, then filtered and washed with EtOAC/IPA (750 mL, 2.5 L/kg, 2:1 v/v with 8 wt% water) followed by IPA (750 mL, 2.5 L/kg). The solids were dried under vacuum at 40 °C to afford Compound 1 hydrochloride salt (170 g, 90% yield). Achiral HPLC purity: 99.91%; chiral HPLC purity: 99.58%. XRPD analysis (FIG.4) confirmed the product (a)
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Inaxaplin (VX-147) is a small-molecule apolipoprotein L1 inhibitor developed by Vertex Pharmaceuticals for APOL1-mediated kidney disease. In preliminary studies the drug has shown promise in treating people with kidney disease and multiple gain of function mutations on the APOL1 gene.[1][2]
FSGS is a disease of the podocyte (glomerular visceral epithelial cells) responsible for proteinuria and progressive decline in kidney function. NDKD is a disease characterized by hypertension and progressive decline in kidney function. Human genetics support a causal role for the G1 and G2 APOL1 variants in inducing kidney disease. Individuals with 2 APOL1 risk alleles are at increased risk of developing primary (idiopathic) FSGS, human immunodeficiency virus (HIV)-associated FSGS, and NDKD. Currently, FSGS and NDKD are managed with symptomatic treatment (including blood pressure control using blockers of the renin angiotensin system), and patients with FSGS and heavy proteinuria may be offered high dose steroids. Corticosteroids induce remission in a minority of patients and are associated with numerous side effects. These patients, in particular individuals of recent sub-Saharan African ancestry with 2 APOL1 risk alleles, experience rapid disease progression leading to end-stage renal disease (ESRD). Thus, there is an unmet medical need for treatment for FSGS and NDKD.
Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
Method G: Amide Coupling with CDMT. A 2 L 3-neck RB flask with magnetic stirrer, temperature probe and nitrogen inlet was charged with 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoic acid S12 (90.5 g, 283.5 mmol) and (3S,4R)-3-amino-4-hydroxy-pyrrolidin-2-one S2 (39.9 g, 343.6 mmol) in DMF (1.65 L), and stirred for 15 minutes. CDMT (61.1 g, 348 mmol) was added. The mixture was then cooled to −2° C. on an ice bath. N-methylmorpholine was added (131 mL, 1.2 mol) dropwise over 20 minutes and the mixture was heated at 30° C. overnight. The reaction mixture was added into approx. 4.5 L of ice water, and extracted with EtOAc (1.2 L×4). The combined organic layers, were washed with 1.2 L of 1 M HCl (×3) and then water (1.2 L) and brine (1.2 L). The combined organic layers were dried over Na 2SO 4, filtered and concentrated. The mixture was washed through a silica gel plug (1.8 L of silica gel), first eluting with 25% EtOAc in dichloromethane (8 L) to remove impurities, followed by hot EtOAc (8 L), to elute the product. The EtOAc filtrate was concentrated in vacuo. TBME was then added (400 mL), and the mixture allowed to stirred for overnight. Filtration of the resulting solid afforded the product as a white solid. 62 g, 52%) 1H NMR (300 MHz, CD 3OD) δ 7.70-7.58 (m, 2H), 7.29-7.13 (m, 3H), 6.73 (ddd, J=11.1, 9.6, 2.2 Hz, 1H), 4.34 (td, J=7.6, 6.8 Hz, 1H), 4.21 (d, J=7.8 Hz, 1H), 3.56 (dd, J=9.9, 7.6 Hz, 1H), 3.20-3.04 (m, 3H), 2.65-2.53 (m, 2H). LCMS m/z 418.2 [M+H] +. Optical rotation: [α] D20.7=−14.01 (c=1.0, 10 mg in 1 mL of MeOH).
Alternative Procedure for Synthesis of Compound (2)
Step 1. Synthesis of Methyl (E)-3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]prop-2-enoate (C51)
A solution of 5,7-difluoro-2-(4-fluorophenyl)-1H-indole C50 (100 g, 1.0 equiv) in dichloromethane (850 mL, 8.5 vol) was agitated at 22° C. Methyl 3,3-dimethoxypropionate (63 mL, 1.1 equiv) was charged followed by trifluoroacetic acid (96 mL, 3.1 equiv), which was rinsed forward with dichloromethane (25 mL, 0.25 vol). The batch was heated to 38° C. and stirred at that temperature. After 4 h, the batch was cooled to 22° C. and charged with n-heptane (200 mL, 2 vol). The mixture was stirred for no less than 1 h at 22° C. The slurry was filtered, and the reactor and the filter cake were washed with n-heptane (1×2 vol (200 mL) and 1×3 vol (300 mL)). The resulting solid was dried under vacuum with nitrogen bleed at 45° C. to afford the product C51 (127.7 g, 95% yield).
Step 2. Synthesis of Methyl 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoate (C52)
To a hydrogenator was charged methyl (E)-3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]prop-2-enoate C51 (100.4 g, 1.0 equiv) followed by Pd(OH) 2/C (0.014 equiv). The vessel was sealed and three vacuum/purge cycles with N2 were performed. 2-MeTHF (2000 mL, 20 vol) was charged using residual vacuum and the resulting mixture was stirred at 22° C. The vessel was sealed and three vacuum/purge cycles with N2 were performed followed by one vacuum purge cycle with hydrogen (H 2). The temperature was adjusted to 22° C., and the vessel pressurized with 20 psi H 2. The mixture was agitated at 22° C. for 4 h. Three vacuum/purge cycles with nitrogen N2 were performed. The batch was filtered through a pad of Hyflo® and the filter cake was rinsed with 2-MeTHF (2×300 mL, 2×3 vol). The combined filtrates were placed under vacuum and distilled at ≤45.0° C. to 2.0 to 3.0 total volumes. The batch temperature was adjusted to 22° C. and the vessel was charge with n-heptane (1000 mL, 10 vol) over at least 1 h. A vacuum was applied and the filtrate distilled at 45.0° C. to 3.5 to 4.5 total volumes. The slurry was cooled to 22° C. and allowed to stir for no less than 1 h. The slurry was filtered and the filter cake was washed with n-heptane (1×1 vol (100 mL) and 1×0.5 vol (50 mL)). The solids were dried under vacuum with nitrogen bleed at 45° C. to afford the product C52 (91.9 g, 91% yield).
Step 3. Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoic Acid (S12)
A mixture of methyl 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]propanoate C52 (80.0 g, 1.0 equiv) and 2-MeTHF (480 mL, 6 vol) was agitated at 22° C. and treated with methanol (72 mL, 0.9 vol). A solution of KOH (27.1 g, 2.0 equiv) in water (107 mL, 1.3 vol) was charged over approximately 20 min. The resulting mixture was heated to an internal temperature of 35° C. and stirred for 3 h. The temperature was adjusted to 22° C. A vacuum was applied and the mixture was distilled at ≤45° C. to 3.0 total volumes. The internal temperature was adjusted to 12° C. The mixture was then charged with water (64 mL, 0.8 vol) and 2-MeTHF (304 mL, 3.8 vol). 6N HCl (75 mL, 0.9 vol) was slowly charged into the mixture with vigorous agitation until the batch attained a pH<3. The internal temperature was adjusted to 22° C., and the biphasic mixture was stirred for no less than 0.5 h. The stirring was stopped and the phases were allowed to separate for no less than 0.5 h. The lower aqueous phase was removed. Water (160 mL, 2 vol) was charged to the reactor at 22° C., and the biphasic mixture stirred for no less than 0.5 h. The stirring was stopped, and the phases allowed separated over no less than 0.5 h. The lower aqueous phase was removed and the batch was filtered through a pad of Hyflo®. The reactor and filter cake were rinsed with 2-MeTHF (160 mL, 2 vol). A vacuum was applied and the combined filtrates distilled at ≤40.0° C. to 2-3 total volumes. The vessel was charged with n-heptane (160 mL, 2 vol), a vacuum was applied and the filtrate distilled at 40.0° C. to 2 total volumes (this step was repeated one additional time). The mixture was then charged with additional n-heptane (144 mL, 1.8 vol). The internal temperature was adjusted to 40° C. and stirred for no less than 2 h. The internal temperature was adjusted to 22° C. over a minimum of 5 h and stirred for no less than 16 hours. The slurry was filtered. The filter cake was washed with n-heptane (3×40 mL, 3×0.5 vol). The solids were dried under vacuum with nitrogen bleed at 45° C. to afford product S12 (72.6 g, 95% yield).
Step 4. Synthesis of 3-[5,7-difluoro-2-(4-fluorophenyl)-1H-indol-3-yl]-N-[(3S,4R)-4-hydroxy-2-oxo-pyrrolidin-3-yl]propanamide (2)
A mixture of S12 (50.0 g, 1.0 equiv), (3S,4R)-3-amino-4-hydroxypyrrolidin-2-one hydrochloride S2 (25.1 g, 1.05 equiv), and CDMT (30.3 g, 1.1 equiv) in DMF (250 mL, 5 vol) was agitated and cooled to 0° C. The reactor was charged with NMM (60 mL, 3.5 equiv) over no less than 1 h, while maintaining the internal temperature at ≤5° C. The batch was stirred at −5° C. for no less than 1 h. The batch was warmed to 22° C. over at least 1 h and stirred at 22° C. for 16 h. The batch was cooled to 0° C. Water (250 mL, 5 vol) was charged, while keeping the internal temperature <20° C. The mixture was charged with a 90/10 mixture of EtOAc/IPA (1000 mL, 20 vol). 6N HCl (40 mL, 0.8 vol) was then charged, while maintaining an internal temperature <10° C., until a pH˜1-3 was achieved. The internal temperature was adjusted to 22° C. and the biphasic mixture stirred for no less than 0.5 h. Stirring was stopped and the phases allowed to separate for no less than 0.5 h. The lower aqueous phase was removed. The aqueous layer was back extracted with a 90/10 mixture of EtOAc/IPA (2×250 mL, 2×5 vol) at 22° C. The combined organic phases from extractions were washed with water (5×500 mL, 5×10 vol) at 22° C., by mixing for no less than 0.5 h and settling for no less than 0.5 h for each wash. The batch was polish filtered. A vacuum was applied and the organic phase distilled at <50° C. to 9.5-10.5 total volumes. The mixture was charged with EtOAc (500 mL, 10 vol), vacuum was applied and the organic phase distilled at <50° C. to 9.5-10.5 total volumes (this step was repeated one more time). The mixture was charged with EtOAc (300 mL, 6 vol) and n-heptane (200 mL, 4 vol). The resulting slurry was heated to 50° C. and stirred for no less than 17 h. The mixture was then cooled to 22° C. over 2 h, and stirred for no less than 1 h. The slurry was filtered. The filter cake was washed with 1:1 EtOAc/n-heptane (2×150 mL, 2×3 vol). The solids were dried under vacuum with nitrogen bleed at ≤45° C. to afford Compound 2 (52.6 g, 80% yield).
Re-Crystallization of Compound 2
Compound 2 (37.6 g, 1.0 equiv) was charged to a reactor followed by a 3:1 mixture of IPA/water (240 mL, 6.4 vol). The slurry was heated to an internal temperature of 75° C. The batch was cooled to an internal temperature of 55° C. and stirred at that temperature for at least 0.5 h. The batch was seeded with 0.5 wt % of a previously generated batch of Compound 2, as a suspension in a mixture of 3:1 IPA/water (4 mL, 0.1 vol). The mixture was stirred at 55° C. for no less than 1.5 h. Water (218 mL, 5.8 vol) was added over minimum period of 5 h while maintaining the temperature at 55° C. The slurry was cooled to 22° C. over no less than 5 h and stirred for no less than 2 h. The slurry was filtered. The filter cake was washed with 2:3 IPA/water (2×114 mL, 2×3 vol). The solids were dried under vacuum with nitrogen bleed at ≤45° C. to afford Compound 2 (34.5 g, 92% yield).
Form A of Compound 2
12.3 kg of Compound 2 was charged to the reactor follow by a 3:1 mixture of 2-propanol/water. Agitation was initiated and the mixture was heated to 75° C. to achieve complete dissolution. The mixture was cooled to 55° C. over 1 hour and agitated at that temperature for 30 minutes. Agitation was continued for 1.5 hours. Water (5.8 vol) was charged over 5 h at 55° C., after which the mixture was cooled to 22° C. over 6 hours. The mixture was agitated at 22° C. for 2 hours then filtered under vacuum. The resulting wet cake was washed with a 3:1 mixture of 2-propanol/water (2.74 vol×2) and pulled dry under vacuum. The wet cake was further dried under vacuum with nitrogen bleed at 45° C. to yield 11.2 kg of Form A.
Hydrate Form A of Compound 2
200 mg of Compound 2 was charged with 10 mL of water. The slurry was cooled to 5° C. and allowed to stir. Hydrate A was observed after 3 days of stirring.
Afimetoran is an immunomodulator and an antagonist of toll-like receptors 7 and 8.1,2 It is also is under investigation in clinical trial NCT04269356 (Study to Assess the Way the Body Absorbs, Distributes, Breaks Down and Eliminates Radioactive BMS-986256 in Healthy Male Participants).
The invention further pertains to pharmaceutical compositions containing at least one compound according to the invention that are useful for the treatment of conditions related to TLR modulation, such as inflammatory and autoimmune diseases, and methods of inhibiting the activity of TLRs in a mammal.
Toll/IL-1 receptor family members are important regulators of inflammation and host resistance. The Toll-like receptor family recognizes molecular patterns derived from infectious organisms including bacteria, fungi, parasites, and viruses (reviewed in Kawai, T. et al., Nature Immunol., 11:373-384 (2010)). Ligand binding to the receptor induces dimerization and recruitment of adaptor molecules to a conserved cytoplasmic motif in the receptor termed the Toll/IL-1 receptor (TIR) domain. With the exception of TLR3, all TLRs recruit the adaptor molecule MyD88. The IL-1 receptor family also contains a cytoplasmic TIR motif and recruits MyD88 upon ligand binding (reviewed in Sims, J.E. et al., Nature Rev. Immunol., 10:89-102 (2010)).
Toll-like receptors (TLRs) are a family of evolutionarily conserved, transmembrane innate immune receptors that participate in the first-line defense. As pattern recognition receptors, the TLRs protect against foreign molecules, activated by pathogen associated molecular patterns (PAMPs), or from damaged tissue, activated by danger associated molecular patterns (DAMPs). A total of 13 TLR family members have been identified, 10 in human, that span either the cell surface or the endosomal compartment. TLR7-9 are among the set that are endosomally located and respond to single-stranded RNA (TLR7and TLR8) or unmethylated single-stranded DNA containing cytosine-phosphate-guanine (CpG) motifs (TLR9).
Activation of TLR7/8/9 can initiate a variety of inflammatory responses (cytokine production, B cell activation and IgG production, Type I interferon response). In the case of autoimmune disorders, the aberrant sustained activation of TLR7/8/9 leads to worsening of disease states. Whereas overexpression of TLR7 in mice has been shown to exacerbate autoimmune disease, knockout of TLR7 in mice was found to be protective against disease in lupus-prone MRL/lpr mice. Dual knockout of TLR7 and 9 showed further enhanced protection.
As numerous conditions may benefit by treatment involving modulation of cytokines, IFN production and B cell activity, it is immediately apparent that new compounds capable of modulating TLR7 and/or TLR8 and/or TLR9 and methods of using these compounds could provide substantial therapeutic benefits to a wide variety of patients.
The present invention relates to a new class of [1,2,4]triazolo[1,5-a]pyridinyl substituted indole compounds found to be effective inhibitors of signaling through TLR7/8/9. These compounds are provided to be useful as pharmaceuticals with desirable stability, bioavailability, therapeutic index, and toxicity values that are important to their drugability.
6-(3-isopropyl-5-(piperidin-4-yl)-1H-indol-2-yl)-7,8-dimethyl-[1,2,4]triazolo[1,5-a]pyrid ine, 2 HCl (47.66 g, 104 mmol), DCE (220 mL), DBU (62.4 mL, 414 mmol), and 2-bromoacetamide (17.14 g, 124 mmol). The reaction flask was capped. The reaction mixture was stirred overnight at room temperature. The reaction mixture was concentrated, diluted with water, and stirred for 30 minutes then filtered. The solid was recrystallized using ethanol to afford 2-(4-(2-(7,8-dimethyl-[1,2,4]triazolo[1,5-a] pyridin-6-yl)-3-isopropyl-1H-indol-5-yl)piperidin-1-yl)acetamide (42.3 g, 93 mmol,
Zevotrelvir (Compound 52) is a coronavirus inhibitor with IC50 ranges of <0.1 μM and <0.1mM for 229E hCoV and SARS-CoV-23C-like (3CL) proteases, respectively. Zevotrelvir has the potential to study viral infections.
Coronaviruses are enveloped, positive-sense, single-stranded RNA viruses. The genomic RNA of CoVs has a 5′-cap structure and 3′-poly-A tail and contains at least 6 open reading frames (ORFs). The first ORF (ORF 1a/b) directly translates two polyproteins: pp1a and pp1ab. These polyproteins are processed by a 3C-Like protease (3CLpro), also known as the main protease (Mpro), into 16 non-structural proteins. These non-structural proteins engage in the production of subgenomic RNAs that encode four structural proteins, namely envelope, membrane, spike, and nucleocapsid proteins, among other accessory proteins. As a result, it is understood that 3C-Like protease has a critical role in the coronavirus life cycle.
3CLpro is a cysteine protease involved in most cleavage events within the precursor polyprotein. Active 3CLpro is a homodimer containing two protomers and features a Cys-His dyad located in between domains I and II.3CLpro is conserved among coronaviruses and several common features are shared among the substrates of 3CLpro in different coronaviruses. As there is no human homolog of 3CLpro, it is an ideal antiviral target. Although compounds have been reported to inhibit 3CLpro activity, they have not been approved as coronavirus therapies. (Refer to
WO 2004101742 A2, US 2005/0143320 Al, US 2006/0014821 Al, US 2009/0137818
Al, WO 2013049382 A2, WO 2013166319 A1, WO2018042343, WO2018023054, WO 2022013684, WO 2021252644, WO2022020711, WO 2022020242, US 11,174,231 B1, US 11,124,497 B1, WO 2005113580, and WO2006061714).
There is a need in the art for novel therapeutic agents that treat, ameliorate or prevent SARS-CoV-2 infection. The present invention provides the process of novel compounds which act in inhibiting or preventing SARS-CoV-2 viral replication and thus are used in the treatment of COVID-19 (see PCT/US21/60247).
Synthesis of substituted spirooxindole and its intermediate has been previously published (Refer to PCT/US21/60247, WO2019086142, WO 2020221811, WO2020221826, J. Med. Chem.2012, 55, 9069). However, the scale-up using previous process is very challenging due to the safety concern associated with certain intermediates, instability of certain intermediates as well as lack of purification process other than column chromatograph. Thus, there is a strong need for developing a safe and efficient processes for the large-scale preparation of these novel substituted spirooxindole derivatives.
Example 15. Preparation of Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF (760 kg, 8V) was added into the reaction at 0 °C (-5~5 °C) followed by compound (j) (63 kg, 1.05 eq) and N-Methylmorpholine (56 kg, 2 eq), HATU
(106 kg, 1.0 eq) and Compound (m-1) (100 kg, 1.0 eq). The reactor was rinsed with DMF (190 kg, 2V) under and warmed up to 25 °C (20~30 °C) and stirred for 5 h (3~6 h) at 25 °C (20~30 °C). After that, additional HATU (0.1 eq) was added and the reaction mixture was stirred for 16-24 h.25% Ammonium hydroxide (38 kg) was added to the reaction mixture at 25 °C (20~30 °C) and stirred for 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was then added to water (5000 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (500 kg, 5 V). The cake was dissolved in ethyl acetate (1350 kg, 15 V) and washed with 10% sodium chloride solution (500 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (660 kg, 5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (137 kg, 2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and the wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
Example 16. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF solution of Compound (m-2) (1 kg, 1.0 eq.) was added to a reactor at around 0-10oC. Compound (l) (600 g, 1.0 eq.), NMM (3.00 eq., 850 g) and HATU (1.00 eq., 1.06 kg) was added to the reactor while maintaining the temperature at 0-10oC; The reaction was warmed to 20±5oC, and stirred for at least 6 hours at 20±5oC. HATU (0.20 eq., 210 g) was added to the reactor at 20±5oC and stirred for at least 6 hours at 20±5oC.25% Ammonium hydroxide (390 g, 1.0 eq) was added to the reaction mixture at 20 °C and stirred for 2 h (1~3 h) at 20 °C. EtOAc (14.0 V) and water (14 V) was added at around 25oC over 20 minutes, and the
solution was stirred for at least 30 min. Aqueous phase was extracted with EtOAc for three times and the organic phase was combined, and washed with 10% aq. NaCl for three times at 20±5oC. The organic phase was concentrated to 6 V then EtOH (7.0 V) was charged. The EtOAc-EtOH solvent swap was repeated for three times and concentrated to 5 V before water (7.0 v) was added at 20±5oC. The mixture was cooled to 0-10oC and stirred for 1 h before being filtered. The filter cake was dissolved in ethyl acetate (15 V) and washed with 10% sodium chloride solution for three times. The organic layer was concentrated to 2-3V at not more than 45℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in about 70-75% yield over two steps.
Example 17. Preparation of (3R,5’S)-1′-(N-methyl-N-(4,6,7-trifluoro-1H-indole-2-carbonyl)-L-leucyl)-2-oxospiro[indoline-3,3′-pyrrolidine]-5′-carboxamide (Compound (n))
DMF (10.0 v) was added to a reactor at 25 °C followed by Compound (l) (4.4 kg, 1.0 eq.), NMM (3.0 eq.) Compound (m-3) (1.0 eq.) and HATU (1.0 eq) at 20-25oC. The reaction mixture was stirred for at least 12 hours at 20-25 °C. Once reaction was complete, aqueous ammonium hydroxide (1.0 eq.) was to the reaction system at 20-25 °C, then stirred for at least 2 hours at 20-25oC. The reaction mixture was then added to water (220 kg, 50V) at 20-30°C over 2 h and the resulting slurry was stirred for 2 h (1~5 h) at 25 °C (20~30 °C). The mixture was filtered and the cake was rinsed with water (22 kg, 5 V). The cake was dissolved in ethyl acetate (135 g, 15 V) and washed with 10% sodium chloride solution (22 kg) for three times. The organic layer was separated to 1.5-2.5V at not more than 45 ℃ under vacuum. The solution was cooled to 25 °C (20~30 °C) and Dichloromethane (5V) was added. The mixture was stirred for 2 h (2~5 h) at 25 °C (20~30 °C) and a slurry was formed. n-Heptane (2V) was added dropwise over 0.5 h (0.5~2 h) at 25 °C (20~30 °C) and stirred for additional 2 h (1~3 h) at 25 °C (20~30 °C). The reaction mixture was filtered and wet cake was rinsed with DCM/heptane (5/2). The wet cake was dried at 50 °C (45~55 °C) for 20 h (15~25 h) to provide Compound (n) as the white solid in 80-85% yield.
Ethyl acetate (630 kg, 10 V) was added into reactor (R1) followed by Compound (n) (70 kg). Make sure the water content was less than 0.20% (w/w). The reaction was cooled to 0 °C (-5 – 5°C) and then triethylamine (89.6 kg) was added followed by trifluoroacetic anhydride (92.4 kg) at 0 °C (-5 – 5°C). The reaction was stirred for 1 h (0.5~2 h) at 0 °C (-5 – 5°C). Once the reaction was complete, the reaction mixture was added slowly to 0.2 N aqueous HCl solution (700 kg) over 1 h at 0 °C (-5~5 °C). The resulting solution was stirred for 30 min at 0 °C (-5~5 °C) and the organic layer was separated.1% aqueous ammonium hydroxide (700 kg) was added to the organic layer and stirred at 20 °C for 30 min (15~25 °C). The organic layer was separated and washed with 10% brine for four times. Then the organic layer was separated and distilled to 2-3 V. Toluene-EtOAc swap was performed until precipitate was observed at 3-4 V. Then Toluene (5-6 V) was added and the slurry was stirred at 50 oC for 2 h. Then the solution was cooled down to 20 oC over 1-2 h and stirred for 10 hr (6~14 hr) at 20 °C (15~25 °C). The reaction mixture was filtered and the wet cake was rinsed with toluene (120 kg, 2V). The wet cake was then dried at 50˚C (45~55 °C) for 48 hr to provide desired compound (o) as a white solid in 80-85% yield.
Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 1) Preparation of tert-butyl (1-(2-amino-5-fluorophenyl)pyridin-3-yl)(methyl)carbamate
2,4-Difluoro-1-nitrobenzene (2.0 g, 12.6 ng/mol) and tert-butyl methyl (pyridin-3-yl)carbamate (2.5 g, 1.0 eq.) were dissolved in DMF (20 mL), and K2C03 ( 2.6 g , 1.5 eq .) was added. The internal temperature was maintained at 60–70 ° C and the mixture was stirred for 2 hours. The completion of the reaction was confirmed by TLC when the reaction solution turned deep yellow. After cooling to room temperature, ethyl acetate (EA)/H20 was added, stirred, and the layers were separated. MgS04 was added to the separated organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was dissolved in EtOH (10 mL) and distilled water (10 mL), and then Na 2 S 2 0 4 (13.0 g, 6 eq.) was added. After stirring for 2 hours while maintaining the internal temperature at 60 to 70 ° C, the completion of the reaction was confirmed by TLC when the yellow color of the reaction solution lightened and became almost colorless. After cooling to room temperature, distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. The filtrate was concentrated under reduced pressure, and the obtained residue was separated by column chromatography (n-Hexane/EA = 3/1) to obtain the title compound (2.0 g, 51. ).
Step 2) Preparation of tert-butyl thiazol-4-ylcarbamate
Thiazole-4-carboxylic acid (5.0 g, 38.8 vol) was dissolved in t-Bu0H (100 mL), and then TEA (8.1 mL, 1.5 eq.) and DPPA (7.1 mL, 1.5 eq.) were added. The internal temperature was maintained at 90–100 ° C, and the mixture was stirred for 3 days. The completion of the reaction was confirmed by TLC. The product was concentrated under reduced pressure, distilled water (50 mL) was added, and the solution was washed with EA (100 mL).
It was extracted twice. MgSQ 4 was added to the organic layer, stirred, dried, and filtered.
After concentrating the filtrate under reduced pressure, the residue was added to a small amount of EA, slurried, and the resulting solid was filtered to obtain the white title compound (4.0 g, 51.5%).
Step 3) Preparation of tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate
Step 2) The tert-Butyl thiazol-4-ylcarbamate (4.0 g, 20.0 ng ol) prepared in the reaction vessel was placed in a reaction vessel and the interior was replaced with nitrogen gas. After dissolving in THF (32 mL), it was cooled to _78 ° C using dry ice— acetone. After cooling, LiHMDS (22.4 mL, 1.5 eq.) was slowly added and the reaction mass was stirred for 30 minutes. 4-Bromo-5-chloro-2-fluorobenzenesulfonyl chloride (6.0 g, 1.0 eq.) was dissolved in THF (10 mL) and slowly added to the reaction mixture. The reaction mass was stirred overnight and the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL). MgS0 4 was added to the organic layer, stirred, dried, and filtered. After concentrating the filtrate under reduced pressure, the residue was crystallized from THF/n-hexane to obtain the title compound (4.4 g, 59.0%).
Step 4) Preparation of tert-butyl (l-(2-((4-(N-(tert-butyloxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)-2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate
Tert-butyl (1-(2-amino-5-fluorophenyl)pyrlidin-3-yl)(methyl)carbamate (0.5 g, 1.1 ng ol) prepared in Step 1) and tert-butyl ((4-bromo-5-chloro-2-fluorophenyl)sulfonyl)(thiazol-4-yl)carbamate (0.9 g, 1.2 eq.) prepared in Step 3) were dissolved in 1,4-dioxane (10 mL). Pd(OAc) 2 (0.03 g, 0.1 eq), rac-BINAP (0.19 g, 0.2 eq.), Cs 2 C0 3 (1.5 g, 3.0 eq.) were added to the reaction solution. After reacting at 120 ° C for 30 minutes using a microwave initiator, the completion of the reaction was confirmed by TLC. Distilled water (50 mL) was added and extracted twice with EA (100 mL).
MgS0 4 was added to the organic layer, stirred, filtered and dried. The filtrate was concentrated under reduced pressure, and the residue was separated by column chromatography (EA/n-Hexane = 1/1). This was repeated twice to obtain the title compound (2.0 g, 88.2%).
Step 5) Preparation of 5-chloro-2-fluoro-4-((4-fluoro-2-(3-(methylamino)pyridin-1-yl)phenyl)amino)-N-(thiazol-4-yl)benzenesulfonamide hydrochloride
Step 4) was prepared by adding 1.25 M HCl in MeOH (15 mL) to tert-butyl (1-(2-((4-(Ν-(tert-butoxycarbonyl)-N-(thiazol-4-yl)sulfamoyl)—2-chloro-5-fluorophenyl)amino)-5-fluorophenyl)pyrlidin-3-yl) (methyl)carbamate (2.0 g, 2.9 µl). The mixture was heated to 40–50 ° C and stirred overnight, and the completion of the reaction was confirmed by TLC. The product was concentrated, and methylene chloride (15 mL) was added to the residue, which was stirred for 1 hour, and the resulting solid was filtered to obtain the title compound (0.9 g, 58.8%).
Palopegteriparatide is a human parathyroid hormone analogue corresponding to amino acid residues 1 – 34 of human parathyroid hormone, to which a methoxy polyethylene glycol (molecular weight: ca. 43,000) is bound via a cleavable linker (pegylation site: S1). Palopegteriparatide is a pegylated synthetic peptide (molecular weight: ca. 48,000) consisting of 34 amino acid residues.
Palopegteriparatide was approved for medical use in the European Union in November 2023,[2] and in the United States in August 2024.[1][5]
Medical uses
Palopegteriparatide is indicated for the treatment of adults with hypoparathyroidism.[1][2]
Adverse effects
The US Food and Drug Administration (FDA) prescription label for palopegteriparatide includes warnings for a potential risk of risk of unintended changes in serum calcium levels related to number of daily injections and total delivered dose, serious hypocalcemia and hypercalcemia (blood calcium levels that are too high), osteosarcoma (a rare bone cancer) based on findings in rats, orthostatic hypotension (dizziness when standing), and a risk of a drug interaction with digoxin (a medicine for certain heart conditions).[5]
History
The effectiveness of palopegteriparatide was evaluated in a 26-week, randomized, double-blind, placebo-controlled trial that enrolled 82 adults with hypoparathyroidism.[5] Prior to randomization, all participants underwent an approximate four-week screening period in which calcium and active vitamin D supplements were adjusted to achieve an albumin-corrected serum calcium concentration between 7.8 and 10.6 mg/dL, a magnesium concentration ≥1.3 mg/dL and below the upper limit of the reference range, and a 25(OH) vitamin D concentration between 20 to 80 ng/mL.[5] During the double-blind period, participants were randomized to either palopegteriparatide (N = 61) or placebo (N= 21), at a starting dose of 18 mcg/day, co-administered with conventional therapy (calcium and active vitamin D).[5] Study drug and conventional therapy were subsequently adjusted according to the albumin-corrected serum calcium levels.[5] At the end of the trial, 69% of the participants in the palopegteriparatide group compared to 5% of the participants in the placebo group were able to maintain their calcium level in the normal range, without needing active vitamin D and high doses of calcium (calcium dose ≤ 600 mg/day).[5]
In September 2023, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Yorvipath, intended for the treatment of chronic hypoparathyroidism in adults.[4][6] The applicant for this medicinal product is Ascendis Pharma Bone Diseases A/S.[4] Palopegteriparatide was approved for medical use in the European Union in November 2023.[2]
^ Jump up to:abcdef“Yorvipath EPAR”. European Medicines Agency. 19 October 2020. Archived from the original on 10 December 2023. Retrieved 11 December 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^“Yorvipath Product information”. Union Register of medicinal products. 20 November 2023. Archived from the original on 26 November 2023. Retrieved 11 December 2023.
^ Jump up to:abc“Yorvipath: Pending EC decision”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023. Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged.
^“EU/3/20/2350”. European Medicines Agency. 15 September 2023. Archived from the original on 24 September 2023. Retrieved 24 September 2023.
^World Health Organization (2021). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 86”. WHO Drug Information. 35 (3). hdl:10665/346562.
^World Health Organization (2023). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 89”. WHO Drug Information. 37 (1). hdl:10665/366661.
Clinical trial number NCT04701203 for “A Trial Investigating the Safety, Tolerability and Efficacy of TransCon PTH Administered Daily in Adults With Hypoparathyroidism (PaTHway)” at ClinicalTrials.gov
To a mixture of (5)-2-(3-morpholino-5-(trifluoromethyl)picolinoyl)-N-2-oxo-5-phenyl-2,3-dihydro-lH-benzo[e] [l,4]diazepin-3-yl)hydrazine-l-carboxamide (1.4 kg, 1 eq.) in DCM (11.2 L) in a flask was charged with 4A-MS (1.4 kg) and stirred at 20±5 °C for 2hrs. Then, it was cooled to 0°C, charged with triethylamine (0.62 Kg, 2.5 eq.) and stirred for 10 min. /^-Toluenesulfonyl chloride (0.7 kg, 1.5 eq.) in DCM (1.4 L) solution was dropwise added to the reaction mixture with maintaining below 5°C and stirred at at 0±5 °C for 5 hrs. The reaction mixture was filtered and washed with DCM (2 X 4.2 L). The filtrate was treated with water (4.2 L) at 0°C and stirred between 0 and 10°C for 5 min. After separation, the organic phase was washed with 5% aqueous NaHCC solution (7 L), water (7 L) and brine (7 L) successively and separated. The DCM layer was concentrated in vacuo at below 30°C to leave ~7L of organic layer. MTBE (7 L) was added to organic layer and concentrated in vacuo to leave ~ 7 L of organic layer (This step was repeated once). The organic layer was charged with water (7 L) and stirred at 20±5 °C for 4 hrs. The solid was filtered and washed with MTBE (3 X 2.1 L) and purified water (2.8 L). The wet cake was stirred with ethyl acetate (7 L) for 12 hrs, charged with n-heptane (14 L) and stirred at 20±5 °C for 5 hrs. The solid was filtered, washed with n-heptane (2 X 2.8 L) and dried under vacuum at ambient temperature to provide the title compound (0.776 kg, 99.6% purity by HPLC, 97.8%
chiral purity by chiral HPLC) as a pale yellowish solid. LC-MS(ESI, m/z): 550.17 [M+H]+;
Example 253 was prepared using a procedure similar to that used to prepare Example 160 where ethyl 3-chloro-5-(trifluoromethyl)picolinate was used in place of methyl 5-bromo-3-fluoropicolinate. ESI-MS m/z: 550.2 [M+H] +.
Example 160 Step c Example 160 was prepared using a procedure similar to that used to prepare Example 152 where methyl 5-cyano-3-morpholinopicolinate was used in place of ethyl 2-morpholino-4-(trifluoromethyl)benzoate. ESI-MS m/z: 507.2 [M+H] +. 1H NMR (400 MHz, DMSO-d 6) δ 3.02-3.04 (m, 4H), 3.71-3.73 (m, 4H), 5.19-5.21 (d, J=8.0 Hz, 1H), 7.26-7.30 (m, 1H), 7.34-7.36 (m, 2H), 7.44-7.55 (m, 5H), 7.65-7.70 (m, 1H), 8.13 (s, 1H), 8.72 (s, 1H), 9.42-9.45 (m, 1H), 10.98 (s, 1H).
Example 1. Preparation of isopropyl ((((R,S)-(2R,3R,4R,5R)-5-(2-amino-6-(methylamino)-9H-purin-9-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)-phenoxy-phosphoryl)-L-alaninate
Step 1. Preparation of ((2R,3R,4R,5R)-3-(benzoyloxy)-5-bromo-4-fluoro-4-methyltetrahydrofuran-2-yl)methyl benzoate (2)
To a solution of (2R)-3,5-di-O-benzoyl-2-fluoro-2-C-methyl-D-ribono-γ-lactone (24.8 g, 66.6 mmol) in dry THF (333 mL), under a nitrogen atmosphere and cooled to −30° C., was added lithium tri-tert-butoxyaluminum hydride (1.0 M in THF, 22.6 mL, 22.6 mmol) dropwise. After completion of the addition the reaction mixture was slowly warmed up to −15° C. over 90 min then EtOAc was added (300 mL) and the mixture was quenched with a saturated aq. NH 4Cl solution (200 mL). The resulting solution was filtered on Celite® and the filtrate was extracted twice with EtOAc. The combined organics were dried (Na 2SO 4), filtered and concentrated. The residue was taken up in dry DCM (225 mL) under a nitrogen atmosphere, cooled to −20° C., then PPh 3 (19.1 g, 72.8 mmol) was added. After 10 min of stirring at −20° C., CBr 4 (26.0 g, 78.4 mmol) was added and the reaction mixture was allowed to slowly warm up to 0° C. over 2 h. The resulting mixture was poured onto a silica gel column and eluted with PE/EtOAc (gradient 100:0 to 80:20). The fractions containing the α-bromofuranoside were collected and concentrated to afford the product 2 (18.1 g, 41.3 mmol, 62% over two steps) as a thick colorless oil.
Step 2. Preparation of (2R,3R,4R,5R)-5-(2-amino-6-chloro-9H-purin-9-yl)-2-(benzoyloxymethyl)-4-fluoro-4-methyltetrahydrofuran-3-yl benzoate (3)
2-Amino-6-chloropurine (2.63 g, 15.5 mmol) was suspended in t-BuOH (54 mL) under a nitrogen atmosphere. The reaction mixture was heated to 30° C. then potassium tert-butoxide (1.69 g, 15.1 mmol) was added. After 45 min a solution of bromofuranoside 2 (2.24 g, 5.12 mmol) dissolved in anhydrous MeCN (6 mL) was added, the reaction mixture was heated to 65° C. for 16 h then cooled down to room temperature. A saturated aq. NH 4Cl solution (70 mL) was added and the resulting solution was extracted with EtOAc (3×60 mL). The combined organics were dried (Na 2SO 4), filtered and concentrated. The residue was purified twice by column chromatography (gradient PE/EtOAc 80:20 to 0:100 then 60:40 to 20:80) to afford the product 3 (1.56 g, 2.96 mmol, 57%) as a white solid.
Step 3. Preparation of (2R,3R,4R,5R)-5-(2-amino-6-(methylamino)-9H-purin-9-yl)-4-fluoro-2-(hydroxymethyl)-4-methyltetrahydrofuran-3-ol (4)
To a solution of compound 3 (575 mg, 1.09 mmol) in MeOH (9 mL) was added methylamine (33% in absolute EtOH, 1.7 mL, 1.81 mmol). The reaction mixture was heated to 85° C. in a sealed tube for 16 h, cooled down to room temperature and concentrated. The residue was purified by column chromatography (gradient DCM/MeOH 100:0 to 85:15) then reverse phase column chromatography (gradient H 2O/MeOH 100:0 to 0:100) to afford the product 4 (286 mg, 0.91 mmol, 84%) as a white solid.
Step 4. Preparation of isopropyl ((((R,S)-(2R,3R,4R,5R)-5-(2-amino-6-(methylamino)-9H-purin-9-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)-phenoxy-phosphoryl)-L-alaninate (5)
To a solution of compound 4 (114 mg, 365 μmol) in dry THF (4 mL), under a nitrogen atmosphere and cooled to 0° C. was added t-butyl magnesium chloride (1.0 M in THF, 0.66 mL, 660 μmol) dropwise over 10 min. The reaction mixture was stirred 15 min at 0° C. then another 15 min at room temperature. The reaction mixture was cooled down to 0° C. then a solution of isopropyl ((R,S)-(pentafluorophenoxy)-phenoxy-phosphoryl)-L-alaninate, Ross, B. S., Reddy, P. G., Zhang, H. R., Rachakonda, S., and Sofia, M. J., J. Org, Chem., (2011), (253 mg, 558 μmol) dissolved in dry THF (1 mL) was added dropwise over 10 min. The reaction mixture was stirred at 0° C. for 30 min followed by 18 h at room temperature then quenched with a saturated aq. NH 4Cl solution (4 mL) and extracted with EtOAc (3×5 mL). The combined organics were dried, filtered (Na 2SO 4) and concentrated. The residue was purified by column chromatography (gradient DCM/MeOH 100:0 to 90:10) then reverse phase column chromatography (gradient H 2O/MeOH 100:0 to 0:100) to afford product 5 (a mixture of diastereomers, 101 mg, 174 μmol, 48%) as a white solid.
1H NMR (300 MHz, CD 3OD) δ 7.83 (s, 0.55H), 7.82 (s, 0.45H), 7.38-7.16 (m, 5H), 6.15 (d, J=18.5 Hz, 0.45H), (d, J=18.8 Hz, 0.55H), 4.99-4.88 (overlapped with H 2O, m, 1H), 4.65-4.36 (m, 3H), 4.25-4.17 (m, 1H), 3.97-3.85 (m, 1H), 3.05 (br s, 3H), 1.32-1.28 (m, 3H), 1.25-1.15 (m, 9H). 19F NMR (282 MHz, CD 3OD) δ −162.8 (s), −163.3 (s). 31P NMR (121 MHz, CD 3OD) δ 4.10 (s), 3.99 (s). MS (ESI) m/z calcd. for C 24H 34FN 7O 7P [M+H] + 582.2; found 582.2.
Zelatriazin (NBI-1065846 or TAK-041) is a small-molecule agonist of GPR139. It was developed for schizophrenia and anhedonia in depression but trials were unsuccessful and its development was discontinued in 2023.[1][2][3][4][5][6][7]
Example 2: (S)-2-(4-oxobenzo[d][l,2,3]triazin-3(4H)-yl)-N-(l-(4-(trifluoromethoxy)phenyl)ethyl)acetamide
[0166] To a vial containing 2-(4-oxobenzo[d][l,2,3]triazin-3(4H)-yl)acetic acid (15 mg, 0.073 mmol), HOBT (15 mg, 0.095 mmol) and EDC (21 mg, 0.110 mmol) was added DMF (244 μΕ). After stirring at RT for 5 min, (S)- 1 -(4-(trifluoromethoxy)phenyl)ethanamine (18 mg, 0.088 mmol) and DIPEA (64, 0.366 mmol) were added. The reaction mixture was
allowed to stir at RT for 1 h then water was added (5 mL). The solid was filtered off and washed with water to yield the title compound as a white solid (20 mg, 71 % yield). XH NMR
Example 2(S)-2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)-N-(1-(4-(trifluoromethoxy)phenyl)ethyl)acetamide
To a vial containing 2-(4-oxobenzo[d][1,2,3]triazin-3(4H)-yl)acetic acid (15 mg, 0.073 mmol), HOBT (15 mg, 0.095 mmol) and EDC (21 mg, 0.110 mmol) was added DMF (244 μL). After stirring at RT for 5 min, (S)-1-(4-(trifluoromethoxy)phenyl)ethanamine (18 mg, 0.088 mmol) and DIPEA (64, 0.366 mmol) were added. The reaction mixture was allowed to stir at RT for 1 h then water was added (5 mL). The solid was filtered off and washed with water to yield the title compound as a white solid (20 mg, 71% yield). 1H NMR (500 MHz, DMSO-d6) δ ppm 1.40 (d, J=6.8 Hz, 3H), 4.98 (quin, J=7.1 Hz, 1H), 5.09 (s, 2H), 7.33 (d, J=7.8 Hz, 2H), 7.44-7.49 (m, 2H), 7.93-7.98 (m, 1H), 8.09-8.15 (m, 1H), 8.21-8.29 (m, 2H), 8.85 (d, J=7.8 Hz, 1H); ESI-MS m/z [M+H]+ 393.9.
[0312] To a stirred mixture of (1R,4S,6S)-5-(tert-butoxycarbonyl)-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropane]-6-carboxylic acid (120 mg, 0.449 mmol, 1.0 eq.) and o-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (204 mg, 0.539 mmol, 1.2 eq.) in DMF (2 mL) was added N-ethyl-N-isopropylpropan-2-amine (348 mg, 2.69 mmol, 6.0 eq.). The mixture was stirred for 10 min at 0 °C, and then (2S)-2-amino-3-[(3S)-2-oxopyrrolidin-3-yl]propanamide hydrochloride (102 mg, 0.494 mmol, 1.1 eq.) was added. The mixture was stirred for 1 h at rt. The crude product was purified by C18 column with CH3CN:Water (0.05% FA). The desired fractions were concentrated under reduced pressure to provide tert-butyl (1R,4S,6S)-6-{[(1S)-1-carbamoyl-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl]carbamoyl}-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropane]-5-carboxylate (120 mg, 60 %) as a white solid. LC-MS (ESI, m/z): 421 [M+H]+.
[0313] To a stirred mixture of tert-butyl (1R,4S,6S)-6-{[(1S)-1-carbamoyl-2-[(3S)-2-oxopyrrolidin-3-yl]ethyl]carbamoyl}-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropane]-5-carboxylate (140 mg, 0.333 mmol, 1.0 eq.) in DCM (1 mL) was added hydrogen chloride (3 mL, 2M in Et2O). The mixture was stirred for 1 h at rt, and then concentrated under reduced pressure to afford (2S)-2-[(1R,4S,6S)-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropan]-6-ylformamido]-3-[(3S)-2-oxopyrrolidin-3-yl]propanamide hydrochloride (110 mg, crude) as a white solid. LC-MS (ESI, m/z): 321 [M+H]+.
[0314] To a stirred mixture of (2S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoic acid (70.7 mg, 0.311 mmol, 1.1 eq.) and o-(7-Azabenzotriazol-1-yl)-N,N,N’,N’-tetramethyluronium hexafluorophosphate (129 mg, 0.340 mmol, 1.2 eq.) in DMF (2 mL) were added N-ethyl-N-isopropylpropan-2-amine (219 mg, 1.69 mmol, 6.0 eq.). The mixture was stirred for 10 min at 0 °C, and then (2S)-2-[(1R,4S,6S)-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropan]-6-ylformamido]-3-[(3S)-2-oxopyrrolidin-3-yl]propanamide hydrochloride (101 mg, 0.283 mmol, 1.0 eq.) was added. The mixture was stirred for 1 h at rt and purified by C18 column with CH3CN/Water (0.05% FA). The desired fractions were concentrated under reduced pressure to provide (2S)-2-[(1R,4S,6S)-5-[(2S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl]-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropan]-6-ylformamido]-3-[(3S)-2-oxopyrrolidin-3-yl]propanamide (90.0 mg, 57 %) as a white solid. LC-MS (ESI, m/z): 530 [M+H]+.
[0315] To a stirred mixture of (2S)-2-[(1R,4S,6S)-5-[(2S)-3,3-dimethyl-2-(2,2,2- trifluoroacetamido)butanoyl]-5-azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropan]-6- ylformamido]-3-[(3S)-2-oxopyrrolidin-3-yl]propanamide (90.0 mg, 0.170 mmol, 1.0 eq.) and pyridine (53.7 mg, 0.680 mmol, 4.0 eq.) in DCM (2 mL) was added trifluoroacetic anhydride (64.2 mg, 0.306 mmol, 1.8 eq.). The mixture was stirred for 1 h at rt. The reaction was quenched with water (10 mL). The mixture was extracted with dichloromethane (3 x 10 mL). The organic layers were combined, washed with brine (2 x 10 mL), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to afford the crude product. The crude product was purified by prep-HPLC with the following conditions (Column: Mobile Phase B: ACN; Flow rate: 25 mL/min; Gradient: 38% B to 68% B in 7 min, 68% B; Wave Length: 254 nm; RT1(min): 5.07) to afford (1R,4S,6S)-N-[(1S)-1-cyano-2-[(3S)-2- oxopyrrolidin-3-yl]ethyl]-5-[(2S)-3,3-dimethyl-2-(2,2,2-trifluoroacetamido)butanoyl]-5- azaspiro[bicyclo[2.2.1]heptane-2,1′-cyclopropane]-6-carboxamide (18.2 mg, 20%) as a white solid. 1H NMR (400 MHz, 8.45-9.03 (m, 1H), 7.30- 7.65 (m, 1H), 4.80-4.98 (m, 1H), 4.42-4.76 (m, 2H), 4.02-4.18 (m, 1H), 3.10-3.30 (m, 2H), 2.30-2.44 (m, 1H), 1.97-2.25 (m, 3H), 1.59-1.97 (m, 5H), 1.40-1.58 (m, 1H), 0.90-1.06 (m, 9H), 0.61-0.83 (m, 2H), 0.21-0.54 (m, 2H). LC-MS (ESI, m/z): 512 [M+H]+.
Huntington’s disease (HD) is a progressive, autosomal dominant neurodegenerative disorder of the brain, having symptoms characterized by involuntary movements, cognitive impairment, and mental deterioration. Death, typically caused by pneumonia or coronary artery disease, usually occurs 13 to 15 years after the onset of symptoms. The prevalence of HD is between three and seven individuals per 100,000 in populations of western European descent. In North America, an estimated 30,000 people have HD, while an additional 200,000 people are at risk of inheriting the disease from an affected parent. The disease is caused by an expansion of uninterrupted trinucleotide CAG repeats in the “mutant” huntingtin (Htt) gene, leading to production of HTT (Htt protein) with an expanded poly-glutamine (polyQ) stretch, also known as a “CAG repeat” sequence. There are no current small molecule therapies targeting the underlying cause of the disease, leaving a high unmet need for medications that can be used for treating or ameliorating HD. Consequently, there remains a need to identify and provide small molecule compounds for treating or ameliorating HD.
REFERENCES [1]. Sydorenko, et al. Preparation of heterocyclic and heteroaryl compounds for treating Huntington’s disease. World Intellectual Property Organization, WO2020005873 A1. 2020-01-02.
20240216369THE USE OF A SPLICING MODULATOR FOR A TREATMENT SLOWING PROGRESSION OF HUNTINGTON’S DISEASE
20240132509HETEROCYCLIC AND HETEROARYL COMPOUNDS FOR TREATING HUNTINGTON’S DISEASE
20230405000TABLET FOR USE IN TREATING HUNTINGTON’S DISEASE AND METHOD OF MAKING THE SAME
ClassAnti-inflammatories; Antibacterials; Antidementias; Antineoplastics; Antiparkinsonians; Neuroprotectants; Small molecules
Mechanism of ActionPeptide hydrolase inhibitors
Phase II/IIIAlzheimer’s disease
Phase IIPeriodontal disorders
PreclinicalParkinson’s disease; Squamous cell cancer
27 Jan 2023COR 388 licensed to Lighthouse Pharmaceuticals in the US
01 Aug 2022Atuzaginstat is available for licensing as of 01 Aug 2022. http://www.quincetx.com
01 Aug 2022Cortexyme is now called Quince Therapeutics
You need to be a logged in or subscribed to view this content
This small molecule is an orally available protease inhibitor targeting the lysine proteases of the periodontal pathogen Porphyromonas gingivalis. Known as gingipains, these proteases penetrate gingival tissue and cause inflammation at the site of periodontitis (O’Brien-Simpson et al., 2009). Periodontitis has been linked epidemiologically to cognitive impairment, and P. gingivalis bacterial lipopolysaccharide has been detected in postmortem brain tissue of people with AD (Poole et al., 2013). Oral P. gingivalis has been called a risk factor for Alzheimer’s disease (Kanagasingam et al., 2020).
Cortexyme’s approach is based on the theory that P. gingivalis invades the brain, where gingipains contribute to Alzheimer’s pathology (see Sabbagh and Decourt, 2022). The company reported elevated gingipain in brain tissue from people with AD, and a correlation between levels of gingipain and tau proteins in postmortem middle temporal gyrus from AD and healthy control tissue. P. gingivalis DNA was detected in postmortem cortices from people with AD and healthy controls, and in CSF of AD patients (Jan 2019 news on Dominy et al., 2019). In the same study, they show that in mice, oral P. gingivalis infection led to the appearance of bacterial DNA in the brain, increased brain Aβ42 production, neuroinflammation, and hippocampal degeneration. The first three findings were reported to be reduced by atuzaginstat; results for hippocampal cell death were not reported.
In preclinical work from other labs, infection with P. gingivalis was reported to worsen AD pathology and cognitive impairment in AD transgenic mice, and to cause neuroinflammation, memory impairment, neurodegeneration, micro- and astrogliosis, increased brain Aβ and phospho-tau, and neurofibrillary tangles in wild-type C57Bl6 mice (Ishida et al., 2017; Ilievski et al., 2018; Ding et al., 2018). For a review of the preclinical literature, see Costa et al., 2021.
In human neurons grown in culture, P. gingivalis infection led to tau phosphorylation and degradation, synapse loss, and cell death (Haditsch et al., 2020).
P. gingivalis is associated with cardiovascular disease. In rabbits, oral infection was reported to increase arterial plaque and levels of the inflammatory marker CRP. Both were reversed by treatment with COR388 (2020 AAIC abstract). In aged dogs with periodontal disease, ninety days of COR388 reduced oral bacterial load and gum pathology (Arastu-Kapur et al., 2020). In addition, older dogs had bacterial antigens and ribosomal RNA in their brains, consistent with systemic infection seen in humans.
Findings
Two Phase 1 trials of atuzaginstat were completed by June 2019. In a single-dose study of 5 to 250 mg capsules in 34 healthy adults, the compound was safe and well-tolerated. A multiple-dose study assessed safety and tolerability in 24 healthy older adults (mean age of 60 years) and nine with AD (mean age 72). According to a company press release and a poster presentation at the 2018 CTAD conference, healthy adults received 25, 50, or 100 mg COR388 or placebo every 12 hours for 10 days; AD patients took 50 mg or placebo every 12 hours for 28 days. The pharmacokinetic profiles of COR388 in AD and controls were reported to be similar. All volunteers with AD had P. gingivalis DNA fragments in their CSF at baseline. COR388 caused no serious adverse reactions, and no one withdrew. Gingipains also were reported to degrade ApoE, and 28 days of treatment with COR388 was claimed to reduce CSF ApoE fragments (2020 AAIC abstract).
A Phase 2/3 trial (GAIN) evaluating a 48-week course of COR388 in 643 people with mild to moderate AD began in April 2019. Participants took either 40 mg, 80 mg, or placebo twice daily. The primary endpoint was to be ADAS-Cog11 score, and the ADCS-ADL was added later as a co-primary functional endpoint. Further outcomes included CDR-SB, MMSE, NPI, the Winterlight Speech Assessment, MRI brain scans, and change in periodontal disease status. Investigators assessed CSF Aβ and tau, plus P. gingivalis DNA and gingipains in CSF, blood, and saliva, before and after treatment. A dental substudy of 228 participants is assessing effects of COR388 on periodontal disease. This trial involves 93 sites in the U.S. and Europe. The U.S. sites are offering a 48-week open-label extension.
According to a presentation at the 2020 CTAD, GAIN was fully enrolled. At baseline, more than 80 percent of participants had CSF Aβ and tau levels consistent with amyloid positivity or an AD diagnosis. All had detectable antibodies to P. gingivalis in their blood. In the dental substudy, 90 percent had periodontal disease. In December 2020, an independent data-monitoring committee recommended continuing the trial after a planned futility analysis of 300 patients treated for six months (press release).
In February 2021, the FDA placed a partial clinical hold on GAIN because of liver abnormalities in some participants (press release). Dosing in the open-label extension was stopped, but the placebo-controlled portion of GAIN continued. Cortexyme characterized the liver effects as reversible and showing no risk of long-term effects.
In October 2021, Cortexyme announced top-line results indicating the trial had missed its co-primary endpoints of ADAS-Cog11 and ADCS-ADL (press release). The company reported a statistically significant 57 percent slowing of decline on the ADAS-Cog11 in a subgroup with detectable saliva P. gingivalis DNA at baseline who took the higher dose; a 42 percent slowing on the lower dose did not reach statistical significance. This prespecified subgroup analysis included 242 participants; it found no effect on the ADCS-ADL. Improvements in ADAS-Cog and other cognitive endpoints correlated with reductions in saliva P. gingivalis DNA, according to data presented at CTAD 2021 in November. The most common treatment-related adverse events were gastrointestinal, occurring in 12 to 15 percent of treated participants. The treatment groups had dose-related liver enzyme elevations greater than three times the upper limit of normal, in 7 and 15 percent of participants on low and high doses, respectively, with bilirubin elevation reported in two participants on the high dose. The elevations occurred mainly in the first six weeks of treatment, and all resolved without long-term effects. Discontinuations due to transaminase elevations numbered one on placebo, and five and 17 in the 40 mg and 80 mg groups, respectively. The overall dropout rate was 25 percent in the placebo group, and 40 percent in atuzaginstat groups. There were five deaths in the high dose arm, and one in the low dose. All were deemed unrelated to drug. There was no evidence of ARIA or other imaging abnormalities.
At CTAD, the company announced plans for a confirmatory trial, pending discussions with regulators. The plan was to test atuzaginstat in people with mild to moderate AD and evidence of P. gingivalis infection, at the lower dose of 40 mg twice daily, reached by titration to minimize liver effects. The company was also planning a trial in Parkinson’s disease to begin in 2022. These trials were never registered.
In September 2021, Cortexyme began a Phase 1 trial of a second-generation lysin-gingipain inhibitor, COR588 (press release). This compound is expected to require only once-daily dosing. Results were expected in May 2022.
In January 2022, the company announced that the FDA had placed a full clinical hold on atuzaginstat due to concerns about liver toxicity (press release). The company said it intended to develop its backup compound, COR588, for Alzheimer’s disease, pending Phase 1 results. In July 2022, Cortexyme announced that COR588 had met safety and tolerability endpoints in a single- and multiple-ascending dose study in healthy adults (press release).
In August 2022, Cortexyme discontinued the gingipain inhibitor program, and offered it for external licensing (press release). The company changed its name to Quince, and its focus to bone disease. In January 2023, Quince put out word that it had sold Cortexyme’s legacy small molecule protease inhibitor portfolio to Lighthouse Pharmaceuticals, a company co-founded by a former Cortexyme CEO (press release).
Example 1. Preparation of (S)-N-(7-amino-2-oxo-1-(2,3,6-trifluorophenoxy)heptan-3- yl)cyclopentanecarboxamide(1)hydrochloride
[0224] To a mixture of compound 1.4 (23.0 g, 67.2 mmol, 1.00 eq) in THF (200 mL) was added NMM (6.79 g, 67.2 mmol, 7.38 mL, 1.00 eq), isobutyl carbonochloridate (9.17 g, 67.2 mmol, 8.82 mL, 1.00 eq), and diazomethane (5.65 g, 134 mmol, 2.00 eq) at -40 °C under N2 (15 psi). The mixture was stirred at 0 °C for 30 min. LCMS showed the reaction was completed. FLO (200 mL) was added to the reaction and extracted with two 300-mL portions of ethyl acetate. The combined organic phase was washed with two 200-mL portions of brine (200, dried with anhydrous Na2SO4, filtered and concentrated under vacuum to provide crude compound 1.3 (30.0 g, crude) as a yellow oil.
[0225] To a mixture of compound 1.3 (20.0 g, 54.6 mmol, 1.00 eq) in EtOAc (300 mL) was
added hydrogen bromide(29.8 g, 121.7 mmol, 20.0 mL, 33% purity, 2.23 eq) at -20 °C under
N2 (15 psi). The mixture was stirred at -20 °C for 10 min. TLC (petroleum ether : ethyl
acetate = 0:1) showed the reaction was completed. The reaction was basified by addition of
saturated NaHCO3 until the pH of the mixture reached 8, and the mixture was extracted with
three 500-mL portions of EtOAc. The combined organic phase was washed with two 200-mL portions of brine, dried over anhydrous Na2SO4, filtered and concentrated under vacuum
to afford crude compound 1.2 (15.0 g, crude) as a yellow solid.
[0226] To a mixture of compound 1.2 (4.00 g, 9.54 mmol, 1.00 eq) in DMF (40.0 mL) was
Vorbipiprant (CR6086) is an EP4 receptor antagonist, serving as a targeted immunomodulator. Thus, Vorbipiprant is also a potential immune checkpoint inhibitor, to turn cold tumors into hot tumors. Vorbipiprant also antagonizes PGE2-stimulated cAMP production (IC50=22 nM). Vorbipiprant exhibit striking DMARD effects in rodents, and anti-inflammatory activity to inhibt immune-mediated inflammatory diseases.
SCHEME
PATENT
Rottapharm S.p.A.
World Intellectual Property Organization, WO2013004290
Example 7: 4-(1-(6-(4-(trifluoromethyl)benzyl)-6-azaspiro[2.5]octane-5-carboxamido)cyclopropyl)benzoic acid (single unknown enantiomer) (E7)
Procedure A:
The title compound (E7) (54 mg) was prepared according to the general procedure for esters hydrolysis (Method B) starting from methyl 4-(1 -(6-(4-(trifluoromethyl)benzyl)-6-azaspiro[2.5]octane-5-carboxamido)cyclopropyl)benzoate (D122b) (100mg). (LiOH: 4 eq; Reaction time: 18 hrs; RT)
methyl 4-(1 -(6-(4-(trifluoromethyl)benzyl)-6-azaspiro[2.5]octane-5-carboxamido)cyclopropyl)benzoate (D123)) (17.7 g, 36.38 mmol) was partitioned between dioxane (485 ml) and water (242 ml) prior addition of LiOH H2O (6.1 g,
145.5 mmol). The mixture was stirred at RT for 10 hrs. Water (200 ml) was added followed by addition of acetic acid (5.27 ml). Dioxane was evaporated off and acetic acid was added until the pH of the aqueous solution reached the value of ~ 4. The white solid was filtered from the reaction and dried under vacuum overnight then 24 hrs under vacuum at 40 °C affording the title compound (E7) (16.7g).
A peptide having the amino acid sequence of SEQ ID NO: 1 (LQVVYLH: SEQ ID NO: 1) was produced by Peptron Inc. Specifically, coupling was performed one by one starting from the C-terminus using the Fmoc SPPS (9-Fluorenylmethyloxycarbonyl solid phase peptide synthesis) method using an automatic synthesizer (ASP48S, Peptron Inc).
NH 2 -His(Trt)-2-chloro-Trityl Resin , in which the first amino acid at the C-terminus of the peptide was attached to the resin, was used. All amino acid raw materials used in peptide synthesis have the N-terminus protected by Fmoc, and all residues are trityl (Trt), t-butyloxycarbonyl (Boc), t-butyl (t-Bu), etc., which are removed by acid. The protected one was used. As a coupling reagent, HBTU (2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate)/HOBt (Hydroxxybenzotriazole)/NMM (N-methylmorpholine) was used. (1) Protected amino acid (8 equivalents) and coupling reagent HBTU (8 equivalents)/HOBt (8 equivalents)/NMM (16 equivalents) were dissolved in DMF (Dimethylformamide) and added, followed by reaction at room temperature for 2 hours. (2) Fmoc removal was performed twice for 5 minutes at room temperature by adding 20% piperidine in DMF. After repeating reactions (1) and (2) to create the basic peptide skeleton, TFA (trifluoroacetic acid)/EDT (1,2-ethanedithiol)/Thioanisole/TIS (triisopropylsilane)/H 2 O=90/ 2.5 / Peptides were separated from the resin using 2.5/2.5/2.5. After purification by reverse phase HPLC using a Vydac Everest C18 column (250 mm × 22 mm, 10 μm), water-acetonitrile linear gradient (10~75% ( v/v) of acetonitrile) method. The molecular weight of the purified peptide was confirmed using LC/MS (Agilent HP1100 series) and lyophilized.
This is a prodrug of homotaurine, a modified amino acid previously developed under the names tramiprosate and Alzhemed. ALZ-801 is converted to homotaurine in vivo, but is more easily absorbed and lasts longer in the blood than tramiprosate.
Tramiprosate was reported to inhibit Aβ42 aggregation into toxic oligomers (Gervais et al., 2007; Kocis et al., 2017). Both ALZ-801 and tramiprosate are metabolized to 3-sulfopranpanoic acid (3-SPA), which is normally found in brain and also inhibits Aβ42 aggregation (Hey et al., 2018). A more recent study found that homotaurine activates GABA receptors, and suggests an alternative mechanism of action for ALZ-801 (Meera et al., 2023).
After tramiprosate failed in Phase 3, its maker, NeuroChem, marketed it as a nutritional supplement. Years later, a subgroup analysis of the trial data indicated a potential positive effect in participants who carried two copies of ApoE4 (Abushakra et al., 2016; Abushakra et al., 2017). Alzheon licensed ALZ-801 from NeuroChem and is developing it for Alzheimer’s disease.
ALZ-801 is a potent and orally available small-molecule β-amyloid (Aβ) anti-oligomer and aggregation inhibitor, valine-conjugated proagent of Tramiprosate with substantially improved PK properties and gastrointestinal tolerability compared with the parent compound. ALZ-801 is an advanced and markedly improved candidate for the treatment of alzheimer’s disease.
General/Typical Procedure: [0311] (i) The solid material was dissolved in water (25 mL). The solution was passed through a Dowex Marathon C ion-exchange column (strongly acidic, 110 g (5 eq), prewashed). The strong acidic fractions were combined and treated with concentrated HCl (10 mL). The mixture was stirred at 50° C. for 30 minutes, and then was concentrated to dryness. The residual material was co-evaporated with EtOH (ethanol) to completely remove water. EtOH (100 mL) was added to the residue. The mixture was stirred at reflux for 1 h, and then cooled to room temperature. The solid material was collected by filtration. The solid material was dissolved in water (10 mL). The solution was added drop wise to EtOH (100 mL). The product slowly crystallized. The suspension was stirred at room temperature for 30 minutes. The solid material was collected by filtration and it was dried in a vacuum oven (60° C.). ID A2. 1H NMR (D2O).δ. 0.87-0.90 (m, 6H), 1.83 (qt, J = 7.2 Hz, 2H), 2.02-2.09 (m, 1H), 2.79 (t, J = 7.8 Hz, 2H), 3.20-3.29 (m, 2H), 3.60 (d, J = 6.3 Hz, 2H); 13C NMR (D2O).δ. 17.20, 17.77, 24.11, 30.00, 38.29, 48.63, 58.96, 169.35; m/z 237 (M-1).
Avenciguat (BI-685509) is a potent and orally active sGC activator. Avenciguat restores cyclic guanosine monophosphate (cGMP) and improves functionality of nitric oxide (NO) pathways. Avenciguat can be used in research of chronic kidney disease (CKD) and diabetic kidney disease (DKD).
Avenciguat is under investigation in clinical trial NCT05282121 (A Study to Test Whether BI 685509 Alone or in Combination With Empagliflozin Helps People With Liver Cirrhosis Caused by Viral Hepatitis or Non-alcoholic Steatohepatitis (NASH) Who Have High Blood Pressure in the Portal Vein (Main Vessel Going to the Liver)).

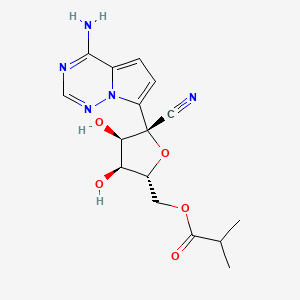








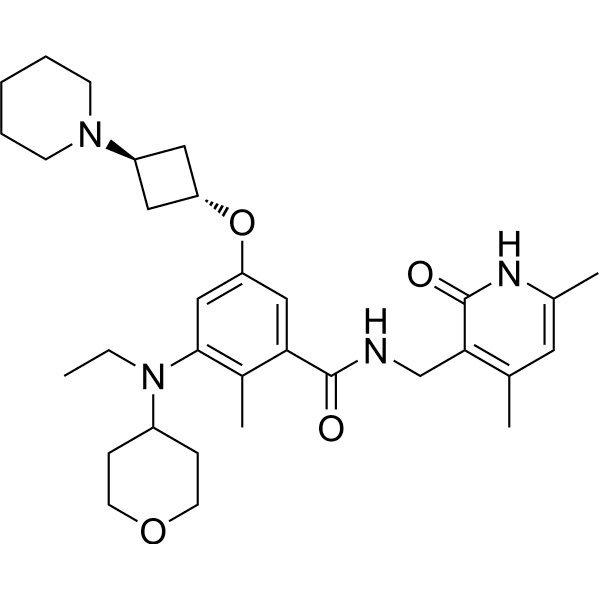
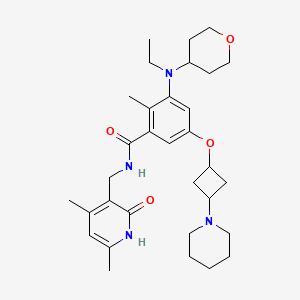

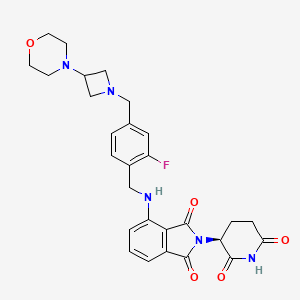




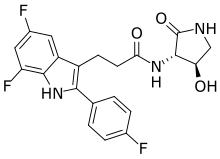



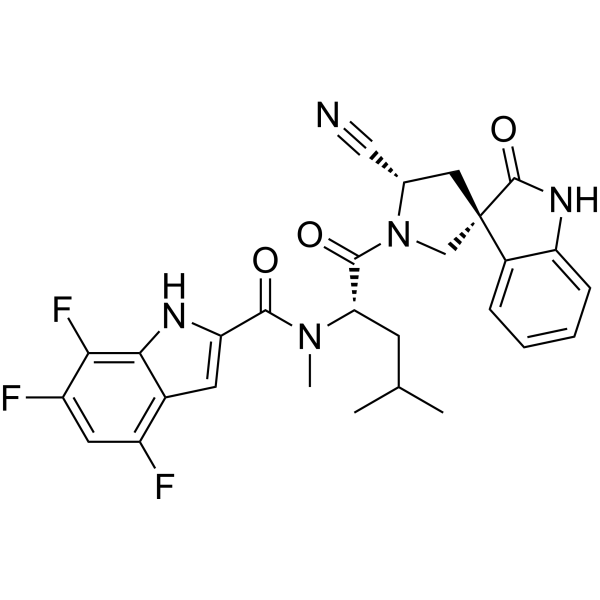
























































 .
.




