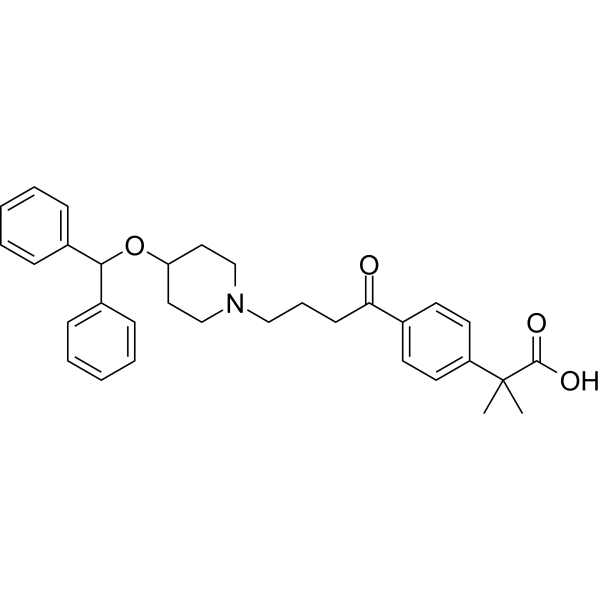
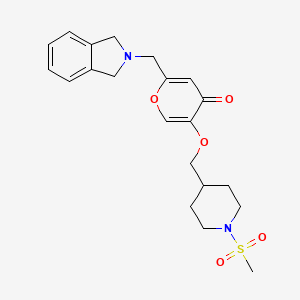
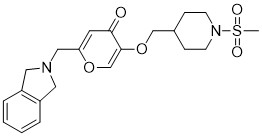
CAS Name: 2-[5-Ethyltetrahydro-5-[tetrahydro-3-methyl-5-[tetrahydro-6-hydroxy-6-(hydroxymethyl)-3,5-dimethyl-2H-pyran-2-yl]-2-furyl]-2-furyl]-9-hydroxy-b-methoxy-a,g,2,8-tetramethyl-1,6-dioxaspiro[4.5]decane-7-butyric acid
Literature References: Polyether antibiotic. Major factor in antibiotic complex isolated from Streptomyces cinnamonensis. Discovery and isolation: Haney, Hoehn, Antimicrob. Agents Chemother.1967, 349. Production: Haney, Hoehn, US3501568 (1970 to Lilly). Structure: Agtarap et al.,J. Am. Chem. Soc.89, 5737 (1967). Crystal structure studies: Lutz et al.,Helv. Chim. Acta53, 1732 (1970); ibid.54, 1103 (1971). Fermentation studies: Stark et al.,Antimicrob. Agents Chemother.1967, 353. Chemistry: Agtarap, Chamberlin, ibid. 359. Stereocontrolled total synthesis: T. Fukuyama et al.,J. Am. Chem. Soc.101, 262 (1979); D. B. Collum et al.,ibid.102, 2117, 2118, 2120 (1980). 13C-NMR study: J. A. Robinson, D. L. Turner, Chem. Commun.1982, 148. Biosynthesis: Day et al.,Antimicrob. Agents Chemother.4, 410 (1973). Review: Stark, “Monensin, A New Biologically Active Compound Produced by a Fermentation Process”, in Fermentation Advances, Pap. Int. Ferment. Symp., 3rd, 1968, D. Perlman, Ed. (Academic Press, New York, 1969) pp 517-540.
Properties: Crystals, mp 103-105° (monohydrate). [a]D +47.7°. pKa 6.6 (in 66% DMF). Very stable under alkaline conditions. Slightly sol in water; more sol in hydrocarbons; very sol in other organic solvents. LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn).
Melting point: mp 103-105° (monohydrate)
pKa: pKa 6.6 (in 66% DMF)
Optical Rotation: [a]D +47.7°
Toxicity data: LD50 of monensin complex in mice, chicks (mg/kg): 43.8 ± 5.2, 284 ± 47 orally (Haney, Hoehn)
The structure of monensin was first described by Agtarap et al. in 1967, and was the first polyether antibiotic to have its structure elucidated in this way. The first total synthesis of monensin was reported in 1979 by Kishi et al.[3]
Production / synthesis Monensin is produced in vivo by Streptomyces cinnamonensis as a natural defense against competing bacteria. Monensin presents a formidable challenge to synthetic chemists as it possesses 17 asymmetric centers on a backbone of only 26 carbon atoms. Although its total synthesis has been described (e.g., Kishi et al., 1979), the high complexity of monensin makes an extraction from the bacterium the most economical procedure for its production. The total synthesis has 56 steps and a yield of only 0.26%. The chemical precursors are 2-allyl-1,3-propanediol and 2- (furan-2-yl)acetonitrile. The method used for synthesizing monensin is based on the principle of “absolute asymmetric synthesis”. Molecules are constructed out of prefabricated building blocks in the correct conformation, aiming for higher yields of the desired enantiomer. New stereocenters are also introduced. Using this method, monensin is assembled in two parts, a larger right side and a smaller left one. The penultimate step is connecting the left and the right halves of monensin, which are independently generated, in an Aldol-condensation. The two halves’ keto end groups (C7/ C8) are linked by eliminating a water molecule. The C7 atom is favored over the C1 atom, because it is more reactive. For catalyzing this step, Yoshito Kishi’s group used iPr2NMgBr (Hauser base) and THF to coordinate it at a temperature of − 78°C. Thus, they were able to isolate the molecule in the right conformation at a ratio of 8:1. Due to the low temperature required for a high yield of the correct enantiomer, the reaction is very solw. One of the most difficult steps is the last one: the connection of the spiro center. This is due to a characteristic feature of spiro compounds; they open and close very easily. Therefore, the conditions for forming the right conformation must be optimal in the last step of synthesis. The biosynthesis in a cell culture of Streptomyces cinnamonensis involves a complex medium containing, among other components, glucose, soybean oil, and grit. Cultivation is carried out for a week at a temperature of 30°C and under constant aeration. Product isolation requires filtration, acidification to pH3, extraction with chloroform and purification with activated carbon. In this way, a few grams per liter of monensin are produced and isolated. For crystallization, azeotropic distillation is necessary. In vivo, polyether backbones are assembled by modular polyketide synthases and are modified by two key enzymes, epoxidase and epoxide hydrolase, to generate the product. Precursors of the polyketide pathway are acetate, butyrate and propionate.
A polyether antibiotic, Monensin was the first member of this class of molecules to be structurally characterized.1 The structural features of these polyethers comprise of a terminal carboxylic acid, multiple cyclic ether rings (ex. Tetrahydrofuran and tetrahydropyran), a large amount of stereocenters and (for many of these molecules) one or more spiroketal moieties.2 Monensin was introduced into the market in 1971 and is used to fight coccidial infections in poultry and as an additive in cattle feed.3 Of the 26 carbon atom’s in Monensin’s backbone, 17 are stereogenic and six of those are contiguous. Coupled with a spiroketal moiety, three hydrofuran rings and two hydropyran rings, the molecule was an attractive synthetic target.
1. Agtarap, A.; Chamberlain, J.W.; Pinkerton, M.; Stein-rauf, L. J. Am. Chem. Soc. 1967, 89, 5737 2. Polyether Antibiotics : Naturally Occurring Acid Ionophores. Westley J.W.; Marcel Dekker: New York (1982) Vol. 1-2. 3. Stark, W.M. In Fermentation Advances, Perlman, D., Ed., Academic Press: New York, 1969, 517
Retrosynthetic Analysis of Monensin
//////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The structure of the sodium (Na+) complex of monensin A.
Monensin A is an ionophore related to the crown ethers with a preference to form complexes with monovalent cations such as: Li+, Na+, K+, Rb+, Ag+, and Tl+.[4][5] Monensin A is able to transport these cations across lipid membranes of cells in an electroneutral (i.e. non-depolarizing) exchange, playing an important role as an Na+/H+antiporter. Recent studies have shown that monensin may transport sodium ion through the membrane in both electrogenic and electroneutral manner.[6] This approach explains ionophoric ability and in consequence antibacterial properties of not only parental monensin, but also its derivatives that do not possess carboxylic groups. It blocks intracellular protein transport, and exhibits antibiotic, antimalarial, and other biological activities.[7] The antibacterial properties of monensin and its derivatives are a result of their ability to transport metal cations through cellular and subcellular membranes.[8]
Uses
Monensin is used extensively in the beef and dairy industries to prevent coccidiosis, increase the production of propionic acid and prevent bloat.[9] Furthermore, monensin, but also its derivatives monensin methyl ester (MME), and particularly monensin decyl ester (MDE) are widely used in ion-selective electrodes.[10][11][12]
In laboratory research, monensin is used extensively to block Golgi transport.[13][14][15]
Toxicity
Monensin has some degree of activity on mammalian cells and thus toxicity is common. This is especially pronounced in horses, where monensin has a median lethal dose 1/100th that of ruminants. Accidental poisoning of equines with monensin is a well-documented occurrence which has resulted in deaths.[16]
^ Kallen, K. J.; Quinn, P.; Allan, D. (1993-02-24). “Monensin inhibits synthesis of plasma membrane sphingomyelin by blocking transport of ceramide through the Golgi: evidence for two sites of sphingomyelin synthesis in BHK cells”. Biochimica et Biophysica Acta (BBA) – Lipids and Lipid Metabolism. 1166 (2–3): 305–308. doi:10.1016/0005-2760(93)90111-l. ISSN0006-3002. PMID8443249.
^ Zhang, G. F.; Driouich, A.; Staehelin, L. A. (December 1996). “Monensin-induced redistribution of enzymes and products from Golgi stacks to swollen vesicles in plant cells”. European Journal of Cell Biology. 71 (4): 332–340. ISSN0171-9335. PMID8980903.
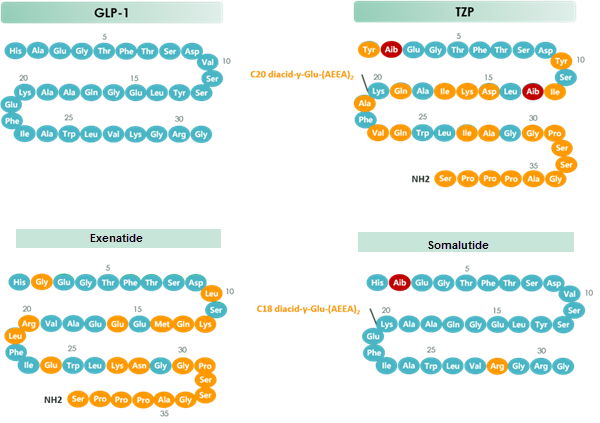
Tirzepatide is an agonist of human glucose-dependent insulinotropic polypeptide (GIP) and human glucagon-like peptide-1 (GLP-1) receptors, whose amino acid residues at positions 2 and 13 are 2-methylAla, and the C-terminus is amidated Ser. A 1,20-icosanedioic acid is attached to Lys at position 20 via a linker which consists of a Glu and two 8-amino-3,6-dioxaoctanoic acids. Tirzepatide is a synthetic peptide consisting of 39 amino acid residues.
Tirzepatide, sold under the brand name Mounjaro,[1] is a medication used for the treatment type 2 diabetes.[2][3][4] Tirzepatide is given by injection under the skin.[2] Common side effects may include nausea, vomiting, diarrhea, decreased appetite, constipation, upper abdominal discomfort and abdominal pain.[2]
The large-scale manufacture of complex synthetic peptides is challenging due to many factors such as manufacturing risk (including failed product specifications) as well as processes that are often low in both yield and overall purity. To overcome these liabilities, a hybrid solid-phase peptide synthesis/liquid-phase peptide synthesis (SPPS/LPPS) approach was developed for the synthesis of tirzepatide. Continuous manufacturing and real-time analytical monitoring ensured the production of high-quality material, while nanofiltration provided intermediate purification without difficult precipitations. Implementation of the strategy worked very well, resulting in a robust process with high yields and purity.
Preclinical, phase I, and phase II trials have indicated that tirzepatide exhibits similar adverse effects to other established GLP-1 receptor agonists, such as GLP-1 receptor agonist dulaglutide. These effects occur largely within the gastrointestinal tract.[5] The most frequently observed adverse effects are nausea, diarrhoea and vomiting, which increased in incidence with the dosage amount (i.e. higher likelihood the higher the dose). The number of patients who discontinued taking tirzepatide also increased as dosage increased, with patients taking 15 mg having a 25% discontinuation rate vs 5.1% for 5 mg patients and 11.1% for dulaglutide.[6] To a slightly lesser extent, patients also reported reduced appetite.[5] Other side effects reported were dyspepsia, constipation, abdominal pain, dizziness and hypoglycaemia.[7][8]
Tirzepatide has a greater affinity to GIP receptors than to GLP-1 receptors, and this dual agonist behaviour has been shown to produce greater reductions of hyperglycemia compared to a selective GLP-1 receptor agonist.[3]Signaling studies have shown that this is due to tirzepatide mimicking the actions of natural GIP at the GIP receptor.[13] However, at the GLP-1 receptor, tirzepatide shows bias towards cAMP (a messenger associated with regulation of glycogen, sugar and lipid metabolism) generation, rather than β-arrestin recruitment. This combination of preference towards GIP receptor and distinct signaling properties at GLP-1 suggest this biased agonism increases insulin secretion.[13] Tirzepatide has also been shown to increase levels of adiponectin, an adipokine involved in the regulation of both glucose and lipid metabolism, with a maximum increase of 26% from baseline after 26 weeks, at the 10 mg dosage.[3]
Chemistry
Structure
Tirzepatide is an analog of the human GIP hormone with a C20 fatty-diacid portion attached, used to optimise the uptake and metabolism of the compound.[9] The fatty-diacid section (eicosanedioic acid) is linked via a glutamic acid and two (2-(2-aminoethoxy)ethoxy)acetic acid units to the side chain of the lysine residue. This arrangement allows for a much longer half life, extending the time between doses, because of its high affinity to albumin.[14]
Synthesis
The synthesis of tirzepatide was first disclosed in patents filed by Eli Lilly and Company.[15] This uses standard solid phase peptide synthesis, with an allyloxycarbonylprotecting group on the lysine at position 20 of the linear chain of amino acids, allowing a final set of chemical transformations in which the sidechain amine of that lysine is derivatized with the lipid-containing fragment.
Large-scale manufacturing processes have been reported for this compound.[16]
History
Indiana-based pharmaceutical company Eli Lilly and Company first applied for a patent for a method of glycemic control using tirzepatide in early 2016.[15] The patent was published late that year. After passing phase 3 clinical trials, Lilly applied for FDA approval in October 2021 with a priority review voucher.[17]
Following the completion of the pivotal SURPASS-2 trial no. NCT03987919, the company announced on 28 April that tirzepatide had successfully met their endpoints in obese and overweight patients without diabetes.[18] Alongside results from the SURMOUNT-1 trial no. NCT04184622, they suggest that tirzepatide may potentially be a competitor for existing diabetic medication semaglutide, manufactured by Novo Nordisk.[19][20]
In industry-funded preliminary trials comparing tirzepatide to the existing diabetes medication semaglutide (an injected analogue of the hormone GLP-1), tirzepatide showed minor improvement of reductions (2.01%–2.30% depending on dosage) in glycated hemoglobin tests relative to semaglutide (1.86%).[21] A 10 mg dose has also been shown to be effective in reducing insulin resistance, with a reduction of around 8% from baseline, measured using HOMA2-IR (computed with fasting insulin).[3] Fasting levels of IGF binding proteins like IGFBP1 and IGFBP2 increased following tirzepatide treatment, increasing insulin sensitivity.[3] A meta-analysis published by Dutta et al. showed that over 1-year clinical use, tirzepatide was observed to be superior to dulaglutide, semaglutide, degludec, and insulin glargine with regards to glycemic efficacy and obesity reduction. Tirzepatide is perhaps the most potent agent developed to date to tackle the global problem of “diabesity“.[22]
Society and culture
Names
Tirzepatide is the international nonproprietary name (INN).[23]
^ Dahl D, Onishi Y, Norwood P, Huh R, Bray R, Patel H, Rodríguez Á (February 2022). “Effect of Subcutaneous Tirzepatide vs Placebo Added to Titrated Insulin Glargine on Glycemic Control in Patients With Type 2 Diabetes: The SURPASS-5 Randomized Clinical Trial”. JAMA. 327 (6): 534–545. doi:10.1001/jama.2022.0078. PMID35133415.
^ Jump up to:abUS patent 9474780, Bokvist BK, Coskun T, Cummins RC, Alsina-Fernandez J, “GIP and GLP-1 co-agonist compounds”, issued 2016-10-25, assigned to Eli Lilly and Co
^ Dutta D, Surana V, Singla R, Aggarwal S, Sharma M (November–December 2021). “Efficacy and safety of novel twincretin tirzepatide a dual GIP and GLP-1 receptor agonist in the management of type-2 diabetes: A Cochrane meta-analysis”. Indian Journal of Endocrinology and Metabolism. 25 (6): 475–489. doi:10.4103/ijem.ijem_423_21.
^World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 81”. WHO Drug Information. 33 (1). hdl:10665/330896.
Frías JP (November 2020). “Tirzepatide: a glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) dual agonist in development for the treatment of type 2 diabetes”. Expert Rev Endocrinol Metab. 15 (6): 379–394. doi:10.1080/17446651.2020.1830759. PMID33030356.
“Tirzepatide”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03954834 for “A Study of Tirzepatide (LY3298176) in Participants With Type 2 Diabetes Not Controlled With Diet and Exercise Alone (SURPASS-1)” at ClinicalTrials.gov
Clinical trial number NCT03987919 for “A Study of Tirzepatide (LY3298176) Versus Semaglutide Once Weekly as Add-on Therapy to Metformin in Participants With Type 2 Diabetes (SURPASS-2)” at ClinicalTrials.gov
Clinical trial number NCT03882970 for “A Study of Tirzepatide (LY3298176) Versus Insulin Degludec in Participants With Type 2 Diabetes (SURPASS-3)” at ClinicalTrials.gov
Clinical trial number NCT03730662 for “A Study of Tirzepatide (LY3298176) Once a Week Versus Insulin Glargine Once a Day in Participants With Type 2 Diabetes and Increased Cardiovascular Risk (SURPASS-4)” at ClinicalTrials.gov
Clinical trial number NCT04039503 for “A Study of Tirzepatide (LY3298176) Versus Placebo in Participants With Type 2 Diabetes Inadequately Controlled on Insulin Glargine With or Without Metformin (SURPASS-5)” at ClinicalTrials.gov
FDA approves Lilly’s Mounjaro (tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes
Mounjaro delivered superior A1C reductions versus all comparators in phase 3 SURPASS clinical trials
While not indicated for weight loss, Mounjaro led to significantly greater weight reductions versus comparators in a key secondary endpoint
Mounjaro represents the first new class of diabetes medicines introduced in nearly a decade and is expected to be available in the U.S. in the coming weeks
INDIANAPOLIS, May 13, 2022 /PRNewswire/ — The U.S. Food and Drug Administration (FDA) approved Mounjaro (tirzepatide) injection, Eli Lilly and Company’s (NYSE: LLY) new once-weekly GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes. Mounjaro has not been studied in patients with a history of pancreatitis and is not indicated for use in patients with type 1 diabetes mellitus.
As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP and GLP-1, which are natural incretin hormones.1
“Mounjaro delivered superior and consistent A1C reductions against all of the comparators throughout the SURPASS program, which was designed to assess Mounjaro’s efficacy and safety in a broad range of adults with type 2 diabetes who could be treated in clinical practice. The approval of Mounjaro is an exciting step forward for people living with type 2 diabetes given the results seen in these clinical trials,” said Juan Pablo Frías, M.D., Medical Director, National Research Institute and Investigator in the SURPASS program.
Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
The approval was based on results from the phase 3 SURPASS program, which included active comparators of injectable semaglutide 1 mg, insulin glargine and insulin degludec. Efficacy was evaluated for Mounjaro 5 mg, 10 mg and 15 mg used alone or in combination with commonly prescribed diabetes medications, including metformin, SGLT2 inhibitors, sulfonylureas and insulin glargine. Participants in the SURPASS program achieved average A1C reductions between 1.8% and 2.1% for Mounjaro 5 mg and between 1.7% and 2.4% for both Mounjaro 10 mg and Mounjaro 15 mg. While not indicated for weight loss, mean change in body weight was a key secondary endpoint in all SURPASS studies. Participants treated with Mounjaro lost between 12 lb. (5 mg) and 25 lb. (15 mg) on average.1
Side effects reported in at least 5% of patients treated with Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion (dyspepsia), and stomach (abdominal) pain. The labeling for Mounjaro contains a Boxed Warning regarding thyroid C-cell tumors. Mounjaro is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with Multiple Endocrine Neoplasia syndrome type 2.1
“Lilly has a nearly 100-year heritage of advancing care for people living with diabetes – never settling for current outcomes. We’re not satisfied knowing that half of the more than 30 million Americans living with type 2 diabetes are not reaching their target blood glucose levels,” said Mike Mason, president, Lilly Diabetes. “We are thrilled to introduce Mounjaro, which represents the first new class of type 2 diabetes medication introduced in almost a decade and embodies our mission to bring innovative new therapies to the diabetes community.”
Mounjaro is expected to be available in the United States in the coming weeks. Lilly is committed to helping people access the medicines they are prescribed and will work with insurers, health systems and providers to help enable patient access to Mounjaro. Lilly plans to offer a Mounjaro savings card for people who qualify. Patients or healthcare professionals with questions about Mounjaro can visit www.Mounjaro.com or call The Lilly Answers Center at 1-800-LillyRx (1-800-545-5979).
Tirzepatide is also under regulatory review for the treatment of type 2 diabetes in Europe, Japan and several additional markets. A multimedia gallery is available on Lilly.com.
About the SURPASS clinical trial program The SURPASS phase 3 global clinical development program for tirzepatide began in late 2018 and included five global registration trials and two regional trials in Japan. These studies ranged from 40 to 52 weeks and evaluated the efficacy and safety of Mounjaro 5 mg, 10 mg and 15 mg as a monotherapy and as an add-on to various standard-of-care medications for type 2 diabetes. The active comparators in the studies were injectable semaglutide 1 mg, insulin glargine and insulin degludec. Collectively, the five global registration trials consistently demonstrated A1C reductions for participants taking Mounjaro across multiple stages of their type 2 diabetes journeys, from an average around five to 13 years of having diabetes.2-8
SURPASS-1 (NCT03954834) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=121), 10 mg (N=121) and 15 mg (N=120) as monotherapy to placebo (N=113) in adults with type 2 diabetes inadequately controlled with diet and exercise alone. From a baseline A1C of 7.9%, Mounjaro reduced participants’ A1C by a mean of 1.8%* (5 mg) and 1.7%* (10 mg and 15 mg) compared to 0.1% for placebo. In a key secondary endpoint, from a baseline weight of 189 lb., Mounjaro reduced participants’ weight by a mean of 14 lb.* (5 mg), 15 lb.* (10 mg) and 17 lb.* (15 mg) compared to 2 lb. for placebo.2,3
SURPASS-2 (NCT03987919) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=470), 10 mg (N=469) and 15 mg (N=469) to injectable semaglutide 1 mg (N=468) in adults with type 2 diabetes inadequately controlled with ≥1500 mg/day metformin alone. From a baseline A1C of 8.3%, Mounjaro reduced participants’ A1C by a mean of 2.0%ꝉ (5 mg), 2.2%* (10 mg) and 2.3%* (15 mg) compared to 1.9% for semaglutide. In a key secondary endpoint, from a baseline weight of 207 lb., Mounjaro reduced participants’ weight by a mean of 17 lb.ꝉ (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to 13 lb. for semaglutide.4,5
SURPASS-3 (NCT03882970) was a 52-week study comparing the efficacy of Mounjaro 5 mg (N=358), 10 mg (N=360) and 15 mg (N=358) to titrated insulin degludec (N=359) in adults with type 2 diabetes treated with metformin with or without an SGLT-2 inhibitor. From a baseline A1C of 8.2%, Mounjaro reduced participants’ A1C by a mean of 1.9%* (5 mg), 2.0%* (10 mg) and 2.1%* (15 mg) compared to 1.3% for insulin degludec. From a baseline weight of 208 lb., Mounjaro reduced participants’ weight by a mean of 15 lb.* (5 mg), 21 lb.* (10 mg) and 25 lb.* (15 mg) compared to an increase of 4 lb. for insulin degludec.6
SURPASS-4 (NCT03730662) was a 104-week study comparing the efficacy and safety of Mounjaro 5 mg (N=328), 10 mg (N=326) and 15 mg (N=337) to insulin glargine (N=998) in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The primary endpoint was measured at 52 weeks. From a baseline A1C of 8.5%, Mounjaro reduced participants’ A1C by a mean of 2.1%* (5 mg), 2.3%* (10 mg) and 2.4%* (15 mg) compared to 1.4% for insulin glargine. From a baseline weight of 199 lb., Mounjaro reduced weight by a mean of 14 lb.* (5 mg), 20 lb.* (10 mg) and 23 lb.* (15 mg) compared to an increase of 4 lb. for insulin glargine.7
SURPASS-5 (NCT04039503) was a 40-week study comparing the efficacy and safety of Mounjaro 5 mg (N=116), 10 mg (N=118) and 15 mg (N=118) to placebo (N=119) in adults with inadequately controlled type 2 diabetes already being treated with insulin glargine, with or without metformin. From a baseline A1C of 8.3%, Mounjaro reduced A1C by a mean of 2.1%* (5 mg), 2.4%* (10 mg) and 2.3%* (15 mg) compared to 0.9% for placebo. From a baseline weight of 210 lb., Mounjaro reduced participants’ weight by a mean of 12 lb.* (5 mg), 17 lb.* (10 mg) and 19 lb.* (15 mg) compared to an increase of 4 lb. for placebo.8
*p<0.001 for superiority vs. placebo or active comparator, adjusted for multiplicity ꝉp<0.05 for superiority vs. semaglutide 1 mg, adjusted for multiplicity
About Mounjaro (tirzepatide) injection1 Mounjaro (tirzepatide) injection is FDA-approved as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus. As the first and only FDA-approved GIP and GLP-1 receptor agonist, Mounjaro is a single molecule that activates the body’s receptors for GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 (glucagon-like peptide-1). Mounjaro will be available in six doses (2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg, 15 mg) and will come in Lilly’s well-established auto-injector pen with a pre-attached, hidden needle that patients do not need to handle or see.
PURPOSE AND SAFETY SUMMARY WITH WARNINGS Important Facts About MounjaroTM (mown-JAHR-OH). It is also known as tirzepatide.
Mounjaro is an injectable prescription medicine for adults with type 2 diabetes used along with diet and exercise to improve blood sugar (glucose).
It is not known if Mounjaro can be used in people who have had inflammation of the pancreas (pancreatitis). Mounjaro is not for use in people with type 1 diabetes. It is not known if Mounjaro is safe and effective for use in children under 18 years of age.
Warnings Mounjaro may cause tumors in the thyroid, including thyroid cancer. Watch for possible symptoms, such as a lump or swelling in the neck, hoarseness, trouble swallowing, or shortness of breath. If you have a symptom, tell your healthcare provider.
Do not use Mounjaro if you or any of your family have ever had a type of thyroid cancer called medullary thyroid carcinoma (MTC).
Do not use Mounjaro if you have Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
Do not use Mounjaro if you are allergic to tirzepatide or any of the ingredients in Mounjaro.
Mounjaro may cause serious side effects, including:
Inflammation of the pancreas (pancreatitis). Stop using Mounjaro and call your healthcare provider right away if you have severe pain in your stomach area (abdomen) that will not go away, with or without vomiting. You may feel the pain from your abdomen to your back.
Low blood sugar (hypoglycemia). Your risk for getting low blood sugar may be higher if you use Mounjaro with another medicine that can cause low blood sugar, such as a sulfonylurea or insulin. Signs and symptoms of low blood sugar may include dizziness or light-headedness, sweating, confusion or drowsiness, headache, blurred vision, slurred speech, shakiness, fast heartbeat, anxiety, irritability, or mood changes, hunger, weakness and feeling jittery.
Serious allergic reactions. Stop using Mounjaro and get medical help right away if you have any symptoms of a serious allergic reaction, including swelling of your face, lips, tongue or throat, problems breathing or swallowing, severe rash or itching, fainting or feeling dizzy, and very rapid heartbeat.
Kidney problems (kidney failure). In people who have kidney problems, diarrhea, nausea, and vomiting may cause a loss of fluids (dehydration), which may cause kidney problems to get worse. It is important for you to drink fluids to help reduce your chance of dehydration.
Severe stomach problems. Stomach problems, sometimes severe, have been reported in people who use Mounjaro. Tell your healthcare provider if you have stomach problems that are severe or will not go away.
Changes in vision. Tell your healthcare provider if you have changes in vision during treatment with Mounjaro.
Gallbladder problems. Gallbladder problems have happened in some people who use Mounjaro. Tell your healthcare provider right away if you get symptoms of gallbladder problems, which may include pain in your upper stomach (abdomen), fever, yellowing of skin or eyes (jaundice), and clay-colored stools.
Common side effects The most common side effects of Mounjaro include nausea, diarrhea, decreased appetite, vomiting, constipation, indigestion, and stomach (abdominal) pain. These are not all the possible side effects of Mounjaro. Talk to your healthcare provider about any side effect that bothers you or doesn’t go away.
Tell your healthcare provider if you have any side effects. You can report side effects at 1-800-FDA-1088 or www.fda.gov/medwatch.
Before using
Your healthcare provider should show you how to use Mounjaro before you use it for the first time.
Before you use Mounjaro, talk to your healthcare provider about low blood sugar and how to manage it.
Review these questions with your healthcare provider:
Do you have other medical conditions, including problems with your pancreas or kidneys, or severe problems with your stomach, such as slowed emptying of your stomach (gastroparesis) or problems digesting food?
Do you take other diabetes medicines, such as insulin or sulfonylureas?
Do you have a history of diabetic retinopathy?
Are you pregnant or plan to become pregnant or breastfeeding or plan to breastfeed? It is not known if Mounjaro will harm your unborn baby.
Do you take birth control pills by mouth? These may not work as well while using Mounjaro. Your healthcare provider may recommend another type of birth control when you start Mounjaro or when you increase your dose.
Do you take any other prescription medicines or over-the-counter drugs, vitamins, or herbal supplements?
How to take
Read the Instructions for Use that come with Mounjaro.
Use Mounjaro exactly as your healthcare provider says.
Mounjaro is injected under the skin (subcutaneously) of your stomach (abdomen), thigh, or upper arm.
Use Mounjaro 1 time each week, at any time of the day.
Do not mix insulin and Mounjaro together in the same injection.
If you take too much Mounjaro, call your healthcare provider or seek medical advice promptly.
Learn more For more information, call 1-800-LillyRx (1-800-545-5979) or go to www.mounjaro.com.
This information does not take the place of talking with your healthcare provider. Be sure to talk to your healthcare provider about Mounjaro and how to take it. Your healthcare provider is the best person to help you decide if Mounjaro is right for you.
MounjaroTM and its delivery device base are trademarks owned or licensed by Eli Lilly and Company, its subsidiaries, or affiliates.
About Lilly Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
This press release contains forward-looking statements (as that term is defined in the Private Securities Litigation Reform Act of 1995) about Mounjaro (tirzepatide 2.5 mg, 5 mg, 7.5 mg, 10 mg, 12.5 mg and 15 mg) injection as a treatment to improve glycemic control in adults with type 2 diabetes, the timeline for supply of Mounjaro to become available, and certain other milestones and ongoing clinical trials of Mounjaro and reflects Lilly’s current beliefs and expectations. However, as with any pharmaceutical product or medical device, there are substantial risks and uncertainties in the process of research, development and commercialization. Among other things, there can be no guarantee that Mounjaro will be commercially successful, that future study results will be consistent with results to date, or that we will meet our anticipated timelines for the commercialization of Mounjaro. For further discussion of these and other risks and uncertainties, see Lilly’s most recent Form 10-K and Form 10-Q filings with the United States Securities and Exchange Commission. Except as required by law, Lilly undertakes no duty to update forward-looking statements to reflect events after the date of this release.
References
Mounjaro. Prescribing Information. Lilly USA, LLC.
Rosenstock, J, et. al. Efficacy and Safety of Once Weekly Tirzepatide, a Dual GIP/GLP-1 Receptor Agonist Versus Placebo as Monotherapy in People with Type 2 Diabetes (SURPASS-1). Abstract 100-OR. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
Rosenstock, J, et. al. (2021). Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. 2021;398(10295):143-155. doi: 10.1016/S0140-6736(21)01324-6.
Frías JP, Davies MJ, Rosenstock J, et al; for the SURPASS-2 Investigators. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. 2021;385(6)(suppl):503-515. doi: 10.1056/NEJMoa2107519
Frias, J.P. Efficacy and Safety of Tirzepatide vs. Semaglutide Once Weekly as Add-On Therapy to Metformin in Patients with Type 2 Diabetes. Abstract 84-LB. Presented virtually at the American Diabetes Association’s 81st Scientific Sessions; June 25-29.
Ludvik B, Giorgino F, Jódar E, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. 2021;398(10300):583-598. doi: 10.1016/S0140-6736(21)01443-4
Del Prato S, Kahn SE, Pavo I, et al; for the SURPASS-4 Investigators. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. 2021;398(10313):1811-1824. doi: 10.1016/S0140-6736(21)02188-7
Dahl D, Onishi Y, Norwood P, et al. Effect of subcutaneous tirzepatide vs placebo added to titrated insulin glargine on glycemic control in patients with type 2 diabetes: the SURPASS-5 randomized clinical trial. JAMA. 2022;327(6):534-545. doi:10.1001/jama.2022.0078
Participants taking tirzepatide lost up to 52 lb. (24 kg) in this 72-week phase 3 study
63% of participants taking tirzepatide 15 mg achieved at least 20% body weight reductions as a key secondary endpoint
INDIANAPOLIS, April 28, 2022 /PRNewswire/ — Tirzepatide (5 mg, 10 mg, 15 mg) achieved superior weight loss compared to placebo at 72 weeks of treatment in topline results from Eli Lilly and Company’s (NYSE: LLY) SURMOUNT-1 clinical trial, with participants losing up to 22.5% (52 lb. or 24 kg) of their body weight for the efficacy estimandi. This study enrolled 2,539 participants and was the first phase 3 global registration trial evaluating the efficacy and safety of tirzepatide in adults with obesity, or overweight with at least one comorbidity, who do not have diabetes. Tirzepatide met both co-primary endpoints of superior mean percent change in body weight from baseline and greater percentage of participants achieving body weight reductions of at least 5% compared to placebo for both estimandsii. The study also achieved all key secondary endpoints at 72 weeks.
For the efficacy estimand, participants taking tirzepatide achieved average weight reductions of 16.0% (35 lb. or 16 kg on 5 mg), 21.4% (49 lb. or 22 kg on 10 mg) and 22.5% (52 lb. or 24 kg on 15 mg), compared to placebo (2.4%, 5 lb. or 2 kg). Additionally, 89% (5 mg) and 96% (10 mg and 15 mg) of people taking tirzepatide achieved at least 5% body weight reductions compared to 28% of those taking placebo.
In a key secondary endpoint, 55% (10 mg) and 63% (15 mg) of people taking tirzepatide achieved at least 20% body weight reductions compared to 1.3% of those taking placebo. In an additional secondary endpoint not controlled for type 1 error, 32% of participants taking tirzepatide 5 mg achieved at least 20% body weight reductions. The mean baseline body weight of participants was 231 lb. (105 kg).
“Obesity is a chronic disease that often does not receive the same standard of care as other conditions, despite its impact on physical, psychological and metabolic health, which can include increased risk of hypertension, heart disease, cancer and decreased survival,” said Louis J. Aronne, MD, FACP, DABOM, director of the Comprehensive Weight Control Center and the Sanford I. Weill Professor of Metabolic Research at Weill Cornell Medicine, obesity expert at NewYork-Presbyterian/Weill Cornell Medical Center and Investigator of SURMOUNT-1. “Tirzepatide delivered impressive body weight reductions in SURMOUNT-1, which could represent an important step forward for helping the patient and physician partnership treat this complex disease.”
For the treatment-regimen estimandiii, results showed:
Average body weight reductions: 15.0% (5 mg), 19.5% (10 mg), 20.9% (15 mg), 3.1% (placebo)
Percentage of participants achieving body weight reductions of ≥5%: 85% (5 mg), 89% (10 mg), 91% (15 mg), 35% (placebo)
Percentage of participants achieving body weight reductions of ≥20%: 30% (5 mg, not controlled for type 1 error), 50% (10 mg), 57% (15 mg), 3.1% (placebo)
The overall safety and tolerability profile of tirzepatide was similar to other incretin-based therapies approved for the treatment of obesity. The most commonly reported adverse events were gastrointestinal-related and generally mild to moderate in severity, usually occurring during the dose escalation period. For those treated with tirzepatide (5 mg, 10 mg and 15 mg, respectively), nausea (24.6%, 33.3%, 31.0%), diarrhea (18.7%, 21.2%, 23.0%), vomiting (8.3%, 10.7%, 12.2%) and constipation (16.8%, 17.1%, 11.7%) were more frequently experienced compared to placebo (9.5% [nausea], 7.3% [diarrhea], 1.7% [vomiting], 5.8% [constipation]).
Treatment discontinuation rates due to adverse events were 4.3% (5 mg), 7.1% (10 mg), 6.2% (15 mg) and 2.6% (placebo). The overall treatment discontinuation rates were 14.3% (5 mg), 16.4% (10 mg), 15.1% (15 mg) and 26.4% (placebo).
Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and the potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
“Tirzepatide is the first investigational medicine to deliver more than 20 percent weight loss on average in a phase 3 study, reinforcing our confidence in its potential to help people living with obesity,” said Jeff Emmick, MD, Ph.D., vice president, product development, Lilly. “Obesity is a chronic disease that requires effective treatment options, and Lilly is working relentlessly to support people with obesity and modernize how this disease is approached. We’re proud to research and develop potentially innovative treatments like tirzepatide, which helped nearly two thirds of participants on the highest dose reduce their body weight by at least 20 percent in SURMOUNT-1.”
Tirzepatide is a novel investigational once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist, representing a new class of medicines being studied for the treatment of obesity. Tirzepatide is a single peptide that activates the body’s receptors for GIP and GLP-1, two natural incretin hormones. Obesity is a chronic, progressive disease caused by disruptions in the mechanisms that control body weight, often leading to an increase in food intake and/or a decrease in energy expenditure. These disruptions are multifactorial and can be related to genetic, developmental, behavioral, environmental and social factors. To learn more, visit Lilly.com/obesity.
Lilly will continue to evaluate the SURMOUNT-1 results, which will be presented at an upcoming medical meeting and submitted to a peer-reviewed journal. Additional studies are ongoing for tirzepatide as a potential treatment for obesity or overweight.
About tirzepatide
Tirzepatide is a once-weekly GIP (glucose-dependent insulinotropic polypeptide) receptor and GLP-1 (glucagon-like peptide-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1 receptor agonists. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with GLP-1 receptor agonism, may result in greater effects on markers of metabolic dysregulation such as body weight, glucose and lipids. Tirzepatide is in phase 3 development for adults with obesity or overweight with weight-related comorbidity and is currently under regulatory review as a treatment for adults with type 2 diabetes. It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH) and heart failure with preserved ejection fraction (HFpEF). Studies of tirzepatide in obstructive sleep apnea (OSA) and in morbidity/mortality in obesity are planned as well.
About SURMOUNT-1 and the SURMOUNT clinical trial program
SURMOUNT-1 (NCT04184622) is a multi-center, randomized, double-blind, parallel, placebo-controlled trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to placebo as an adjunct to a reduced-calorie diet and increased physical activity in adults without type 2 diabetes who have obesity, or overweight with at least one of the following comorbidities: hypertension, dyslipidemia, obstructive sleep apnea or cardiovascular disease. The trial randomized 2,539 participants across the U.S., Argentina, Brazil, China, India, Japan, Mexico, Russia and Taiwan in a 1:1:1:1 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or placebo. The co-primary objectives of the study were to demonstrate that tirzepatide 10 mg and/or 15 mg is superior in percentage of body weight reductions from baseline and percentage of participants achieving ≥5% body weight reduction at 72 weeks compared to placebo. Participants who had pre-diabetes at study commencement will remain enrolled in SURMOUNT-1 for an additional 104 weeks of treatment following the initial 72-week completion date to evaluate the impact on body weight and potential differences in progression to type 2 diabetes at three years of treatment with tirzepatide compared to placebo.
All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg).
The SURMOUNT phase 3 global clinical development program for tirzepatide began in late 2019 and has enrolled more than 5,000 people with obesity or overweight across six clinical trials, four of which are global studies. Results from SURMOUNT-2, -3, and -4 are anticipated in 2023.
About Lilly
Lilly unites caring with discovery to create medicines that make life better for people around the world. We’ve been pioneering life-changing discoveries for nearly 150 years, and today our medicines help more than 47 million people across the globe. Harnessing the power of biotechnology, chemistry and genetic medicine, our scientists are urgently advancing new discoveries to solve some of the world’s most significant health challenges, redefining diabetes care, treating obesity and curtailing its most devastating long-term effects, advancing the fight against Alzheimer’s disease, providing solutions to some of the most debilitating immune system disorders, and transforming the most difficult-to-treat cancers into manageable diseases. With each step toward a healthier world, we’re motivated by one thing: making life better for millions more people. That includes delivering innovative clinical trials that reflect the diversity of our world and working to ensure our medicines are accessible and affordable. To learn more, visit Lilly.com and Lilly.com/newsroom or follow us on Facebook, Instagram, Twitter and LinkedIn. P-LLY
Tirzepatide results superior A1C and body weight reductions compared to insulin glargine in adults with type 2 diabetes
Newly published data show that participants maintained A1C and weight control up to two years in SURPASS-4, the largest and longest SURPASS trial completed to dateNo increased cardiovascular risk identified with tirzepatide; hazard ratio of 0.74 observed for MACE-4 events
SURPASS-4 is the largest and longest clinical trial completed to date of the phase 3 program studying tirzepatide as a potential treatment for type 2 diabetes. The primary endpoint was measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE) to assess CV risk. In newly published data from the treatment period after 52 weeks, participants taking tirzepatide maintained A1C and weight control for up to two years.
The overall safety profile of tirzepatide, assessed over the full study period, was consistent with the safety results measured at 52 weeks, with no new findings up to 104 weeks. Gastrointestinal side effects were the most commonly reported adverse events, usually occurring during the escalation period and then decreasing over time.
“We are encouraged by the continued A1C and weight control that participants experienced past the initial 52 week treatment period and up to two years as we continue to explore the potential impact of tirzepatide for the treatment of type 2 diabetes,” said John Doupis, M.D., Ph.D., Director, Diabetes Division and Clinical Research Center, Iatriko Paleou Falirou Medical Center, Athens, Greece and Senior Investigator for SURPASS-4.
Tirzepatide is a novel investigational once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single molecule, representing a new class of medicines being studied for the treatment of type 2 diabetes.
SURPASS-4 was an open-label global trial comparing the safety and efficacy of three tirzepatide doses (5 mg, 10 mg and 15 mg) to titrated insulin glargine in 2,002 adults with type 2 diabetes with increased CV risk who were treated with between one and three oral antihyperglycemic medicines (metformin, a sulfonylurea or an SGLT-2 inhibitor). Of the total participants randomized, 1,819 (91%) completed the primary 52-week visit and 1,706 (85%) completed the study on treatment. The median study duration was 85 weeks and 202 participants (10%) completed two years.
Study participants had a mean duration of diabetes of 11.8 years, a baseline A1C of 8.52 percent and a baseline weight of 90.3 kg. More than 85 percent of participants had a history of cardiovascular events. In the insulin glargine arm, the insulin dose was titrated following a treat-to-target algorithm with the goal of fasting blood glucose below 100 mg/dL. The starting dose of insulin glargine was 10 units per day, and the mean dose of insulin glargine at 52 weeks was 43.5 units per day.
About tirzepatide Tirzepatide is a once-weekly dual glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) receptor agonist that integrates the actions of both incretins into a single novel molecule. GIP is a hormone that may complement the effects of GLP-1. In preclinical models, GIP has been shown to decrease food intake and increase energy expenditure therefore resulting in weight reductions, and when combined with a GLP-1 receptor agonist, may result in greater effects on glucose and body weight. Tirzepatide is in phase 3 development for blood glucose management in adults with type 2 diabetes, for chronic weight management and heart failure with preserved ejection fraction (HFpEF). It is also being studied as a potential treatment for non-alcoholic steatohepatitis (NASH).
About SURPASS-4 and the SURPASS clinical trial program SURPASS-4 (NCT03730662) is a randomized, parallel, open-label trial comparing the efficacy and safety of tirzepatide 5 mg, 10 mg and 15 mg to insulin glargine in adults with type 2 diabetes inadequately controlled with at least one and up to three oral antihyperglycemic medications (metformin, sulfonylureas or SGLT-2 inhibitors), who have increased cardiovascular (CV) risk. The trial randomized 2,002 study participants in a 1:1:1:3 ratio to receive either tirzepatide 5 mg, 10 mg or 15 mg or insulin glargine. Participants were located in the European Union, North America (Canada and the United States), Australia, Israel, Taiwan and Latin America (Brazil, Argentina and Mexico). The primary objective of the study was to demonstrate that tirzepatide (10 mg and/or 15 mg) is non-inferior to insulin glargine for change from baseline A1C at 52 weeks in people with type 2 diabetes and increased CV risk. The primary and key secondary endpoints were measured at 52 weeks, with participants continuing treatment up to 104 weeks or until study completion. The completion of the study was triggered by the accrual of major adverse cardiovascular events (MACE). Study participants enrolled had to have a mean baseline A1C between 7.5 percent and 10.5 percent and a BMI greater than or equal to 25 kg/m2 at baseline. All participants in the tirzepatide treatment arms started the study at a dose of tirzepatide 2.5 mg once-weekly and then increased the dose in a step-wise approach at four-week intervals to their final randomized maintenance dose of 5 mg (via a 2.5 mg step), 10 mg (via steps at 2.5 mg, 5 mg and 7.5 mg) or 15 mg (via steps at 2.5 mg, 5 mg, 7.5 mg, 10 mg and 12.5 mg). All participants in the titrated insulin glargine treatment arm started with a baseline dose of 10 units per day and titrated following a treat-to-target algorithm to reach a fasting blood glucose below 100 mg/dL.
The SURPASS phase 3 global clinical development program for tirzepatide has enrolled more than 20,000 people with type 2 diabetes across 10 clinical trials, five of which are global registration studies. The program began in late 2018, and all five global registration trials have been completed.
About Diabetes
Approximately 34 million Americans2 (just over 1 in 10) and an estimated 463 million adults worldwide3 have diabetes. Type 2 diabetes is the most common type internationally, accounting for an estimated 90 to 95 percent of all diabetes cases in the United States alone2. Diabetes is a chronic disease that occurs when the body does not properly produce or use the hormone insulin.
Atosiban, sold under the brand name Tractocile among others, is an inhibitor of the hormones oxytocin and vasopressin. It is used as an intravenousmedication as a labour repressant (tocolytic) to halt premature labor. It was developed by Ferring Pharmaceuticals in Sweden and first reported in the literature in 1985.[5] Originally marketed by Ferring Pharmaceuticals, it is licensed in proprietary and generic forms for the delay of imminent preterm birth in pregnant adult women.
Atosiban is an inhibitor of the hormones oxytocin and vasopressin. It is used intravenously to halt premature labor. Although initial studies suggested it could be used as a nasal spray and hence would not require hospital admission, it is not used in that form. Atobisan was developed by the Swedish company Ferring Pharmaceuticals. It was first reported in the literature in 1985. Atosiban is licensed in proprietary and generic forms for the delay of imminent pre-term birth in pregnant adult women.
Medical uses
Atosiban is used to delay birth in adult women who are 24 to 33 weeks pregnant, when they show signs that they may give birth pre-term (prematurely).[4] These signs include regular contractions lasting at least 30 seconds at a rate of at least four every 30 minutes,[4] and dilation of the cervix (the neck of the womb) of 1 to 3 cm and an effacement (a measure of the thinness of the cervix) of 50% or more.[4] In addition, the baby must have a normal heart rate.[4]
Pharmacology
Mechanism of action
Atosiban is a nonapeptide, desamino-oxytocin analogue, and a competitive vasopressin/oxytocin receptor antagonist (VOTra). Atosiban inhibits the oxytocin-mediated release of inositol trisphosphate from the myometrial cell membrane. As a result, reduced release of intracellular, stored calcium from the sarcoplasmic reticulum of myometrial cells and reduced influx of Ca2+ from the extracellular space through voltage-gated channels occur. In addition, atosiban suppresses oxytocin-mediated release of PGE and PGF from the decidua.[6]
In human preterm labour, atosiban, at the recommended dosage, antagonises uterine contractions and induces uterine quiescence. The onset of uterus relaxation following atosiban is rapid, uterine contractions being significantly reduced within 10 minutes to achieve stable uterine quiescence.
Other uses
Atosiban use after assisted reproduction
Atosiban is useful in improving the pregnancy outcome of in vitro fertilization-embryo transfer (IVF-ET) in patients with repeated implantation failure.[7] The pregnancy rate improved from zero to 43.7%.[8]
First- and second-trimester bleeding was more prevalent in ART than in spontaneous pregnancies. From 2004 to 2010, 33 first-trimester pregnancies with vaginal bleeding after ART with evident uterine contractions, when using atosiban and/or ritodrine, no preterm delivery occurred before 30 weeks.[9]
In a 2010 meta-analysis,[10] nifedipine is superior to β2 adrenergic receptor agonists and magnesium sulfate for tocolysis in women with preterm labor (20–36 weeks), but it has been assigned to pregnancy category C by the U.S. Food and Drug Administration, so is not recommended before 20 weeks, or in the first trimester.[9] A report from 2011 supports the use of atosiban, even at very early pregnancy, to decrease the frequency of uterine contractions to enhance success of pregnancy.[7]
Pharmacovigilance
Following the launch of atosiban in 2000, the calculated cumulative patient exposure to atosiban (January 2000 to December 2005) is estimated as 156,468 treatment cycles. To date, routine monitoring of drug safety has revealed no major safety issues.[11]
Regulatory affairs
Atosiban was approved in the European Union in January 2000 and launched in the European Union in April 2000.[12][4] As of June 2007, atosiban was approved in 67 countries, excluding the United States and Japan.[12] It was understood that Ferring did not expect to seek approval for atosiban in the US or Japan, focusing instead on development of new compounds for use in Spontaneous Preterm Labor (SPTL).[12] The fact that atosiban only had a short duration before it was out of patent that the parent drug company decided not to pursue licensing in the US.[13]
Systematic reviews
In a systematic review of atosiban for tocolysis in preterm labour, six clinical studies — two compared atosiban to placebo and four atosiban to a β agonist — showed a significant increase in the proportion of women undelivered by 48 hours in women receiving atosiban compared to placebo. When compared with β agonists, atosiban increased the proportion of women undelivered by 48 hours and was safer compared to β agonists. Therefore, oxytocin antagonists appear to be effective and safe for tocolysis in preterm labour.[14]
A 2014 systematic review by the Cochrane Collaboration showed that while atosiban had fewer side effects than alternative drugs (such as ritodrine), other beta blockers, and calcium channel antagonists, it was no better than placebo in the major outcomes i.e. pregnancy prolongation or neonatal outcomes. The finding of an increase in infant deaths in one placebo-controlled trial warrants caution. Further research is recommended.[15]
PATENT
WO 2021207870
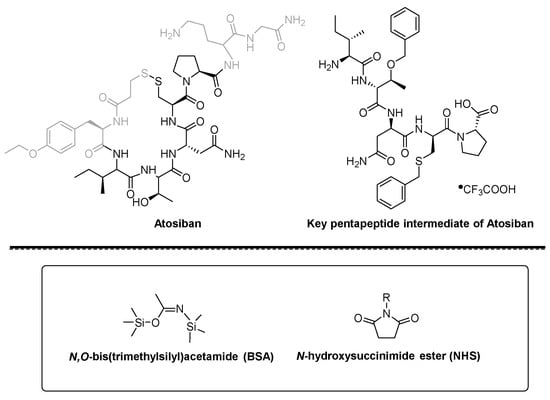
Atosiban (Atosiban) is an oxytocin and vasopressin V1A combined receptor antagonist, which can be used as a competitive antagonist of cyclic peptide oxytocin receptors in the uterus, decidua and fetal membrane. Atosiban is a disulfide-bonded cyclic polypeptide composed of 9 amino acids. It is a modified oxytocin molecule at positions 1, 2, 4 and 8. The N-terminal of the peptide is 3-mercaptopropionic acid (thiol and [ Cys] 6 thiol forms a disulfide bond), the C-terminal is in the form of an amide, and the second amino acid at the N-terminal is ethylated [D-Tyr(Et)] 2 . Atosiban is generally present in medicines in the form of acetate salt, commonly known as atosiban acetate. Its chemical formula is C 45 H 71 N 11 O 14 S 2 , its molecular weight is 994.19, and its structural formula is as follows:
[0003]
[0004]
In the prior art, atosiban is usually synthesized by a solid-phase peptide synthesis (SPPS) method, an amino resin is used as a starting carrier resin, and protected amino acids are sequentially connected, and the obtained atosiban is oxidized and then cleaved to obtain atosiban. However, the above-mentioned existing process has high cost, generates a large amount of solvent waste, and is not easy to monitor during the cyclization process. In addition, the above-mentioned prior art has deficiencies in the overall yield of crude peptides. Moreover, due to the existence of D-Tyr(Et) in the structure of atosiban, Fmoc-D-Tyr(Et) easily undergoes a racemization reaction during the peptide attachment process, resulting in [Tyr(Et) 2 ]-A The impurity of tosiban, which is similar in polarity to atosiban itself, is difficult to completely remove through purification, thus affecting the quality of atosiban.
[table 0001]
Amino acid name
alphabetic symbols
Glycine
Gly
Ornithine
Orn
Proline
Pro
cysteine
Cys
Asparagine
Asn
Threonine
Thr
Isoleucine
Ile
D-tyrosine (oxyethyl)
D-Tyr(ET)
Table 3 List of intermediates and Fmoc protected amino acids
According to the most preferred embodiment of the present invention, the method of the present invention comprises the following steps:
[0046]
The first step: Fmoc-Gly Rink resin can be directly purchased, which reduces the first step of synthesis and improves the synthesis efficiency;
[0047]
The second step: preparing a deprotection solution: the deprotection solution is a mixture of piperidine/N,N-dimethylformamide, preferably piperidine/N,N-dimethylformamide in a volume ratio of 1/4.
[0048]
The third step: preparation of Fmoc-Orn(Boc)-Gly Rink resin: deprotect the Fmoc-Gly Rink resin obtained in the first step, wash with DMF, add Fmoc-Orn(Boc)-OH in DMF solution, Condensation reaction is carried out under the condition of peptide coupling condensing agent to obtain Fmoc-Orn(Boc)-Gly Rink resin;
[0049]
The fourth step: preparation of Fmoc-Pro-Orn(Boc)-Gly Rink resin: the peptide resin obtained in the fourth step is deprotected and washed, and then reacted with Fmoc-Pro-OH under the condition of a peptide coupling agent to obtain Fmoc-Pro-Orn(Boc)-Gly Rink resin;
[0050]
The fifth step: preparation of Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the fifth step is deprotected and washed, and then reacted with Fmoc-Cys(Trt)-OH under the condition of peptide coupling agent to obtain Fmoc-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0051]
The sixth step: preparation of Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the sixth step is deprotected and washed, and then reacted with Fmoc-Asn-OH under the condition of peptide coupling agent to obtain Fmoc-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin ;
[0052]
The seventh step: preparation of Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the seventh step was deprotected and washed, and then reacted with Fmoc-Thr(tBu)-OH under the condition of a peptide coupling agent. Obtain Fmoc-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0053]
The eighth step: preparation of Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the eighth step is deprotected and washed, and then reacted with Fmoc-Ile-OH under the condition of a peptide coupling agent to obtain Fmoc-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn (Boc)-Gly Rink resin;
[0054]
The ninth step: preparation of Fmoc-D-Tyr(RT)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the ninth step is deprotected and washed, and then reacted with Fmoc-D-Tyr(ET)-OH under the condition of a peptide coupling agent to obtain Fmoc-D-Tyr(RT)-Ile-Thr(tBu )-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0055]
The tenth step: preparation of Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin. The peptide resin obtained in the tenth step is deprotected and washed, and then reacted with Mpa(Trt) under the condition of a peptide coupling agent to obtain Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn -Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin;
[0056]
The eleventh step: Mpa(Trt)-D-Tyr(ET)-Ile-Thr(tBu)-Asn-Cys(Trt)-Pro-Orn(Boc)-Gly Rink resin in TFA/TIS/EDT/H2O =90/54/10/5 TFA, cleaved for 3 hours, and filtered to obtain crude peptide solution;
[0057]
The twelfth step: sedimentation and washing of the crude peptide solution with methyl tert-butyl ether, centrifugation at 2000 rpm, and vacuum drying to obtain a pale yellow solid powder of atosiban linear crude peptide;
[0058]
The thirteenth step: prepare three solutions for atosiban cyclization: solution A-sodium acetate buffered aqueous solution, solution B-aqueous solution of linear peptide atosiban crude peptide acetic acid, solution C: 30%-60% hydrogen peroxide solution ;
[0059]
The fourteenth step: Mix the above three solutions of A, B, and C at 15-25 ° C, and stir for 1-3 hours after mixing, so that the Mpa at the 1st position and the Cys at the 6th position form a disulfide bond to obtain Cyclized atosiban crude peptide.
[0060]
Step fifteen: Purify crude atosiban by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes.
[0061]
The sixteenth step: freeze-dry the purified atosiban solution at -50 to -70° C. for 18-48 hours with a freeze dryer.
[0062]
The purity of atosiban obtained by the method of the invention is more than 99.5%, and the total product yield is 55%-65%.
[0063]
The advantage of the method for preparing atosiban of the present invention is:
[0064]
The traditional SPPS synthesis of atosiban usually produces a large amount of waste with high disposal costs. This process adopts high-temperature SPPS process and selects different condensing agent combinations, which is faster than the conventional SPPS process, the product purity can reach more than 99.9%, the purity is better than that of the conventional atosiban process, the impurity content is low, and the product quality is high. The total yield can reach 55%-65%.
Detailed ways
[0065]
The invention will now be described with reference to specific embodiments. It must be understood that these examples are merely illustrative of the invention and are not intended to limit the scope of the invention. Unless otherwise stated, percentages and parts are by weight. Unless otherwise specified, experimental materials and reagents used in the following examples were obtained from commercial sources.
[0066]
Example 1:
[0067]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.61 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, other amino acids in the sequence are connected in the following order, and the coupling reagents are N,N-diisopropylcarbodiimide, 2-(7-benzotriazole)-N,N,N’,N ‘-Tetramethylurea hexafluorophosphate mixed in a volume ratio of 1:1, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 90°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated and washed twice with methyl tert-butyl ether, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 2:2 A solution-acetic acid-sodium acetate buffer aqueous solution (concentration is 30g/L), B solution-linear peptide atosiban crude peptide acetic acid aqueous solution and C solution: 60% hydrogen peroxide solution.
[0068]
The crude peptide yield was 85%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 18 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 56%.
[0069]
Example 2:
[0070]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, the other amino acids in the sequence are connected in the following order, and the coupling reagents are N,N-tert-dicyclohexylcarbodiimide, 1-hydroxybenzotriazole and Oxyma, which are mixed in a volume ratio of 1:1:1 , Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D- Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 90°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated with methyl tert-butyl ether and washed twice, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 3:2 solution A-formic acid-sodium formate buffer aqueous solution (concentration 25g/L), solution B-linear peptide atosiban crude peptide formic acid aqueous solution and solution C: 30% hydrogen peroxide solution, and oxygen was introduced.
[0071]
The crude peptide yield was 83%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 18 hours with a freeze dryer, the obtained atosiban has a purity greater than 99.5%, and the total product yield is 57%.
[0072]
Example 3:
[0073]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:4 v/v). Subsequently, other amino acids in the sequence were connected in the following order, and the coupling reagents were N,N-diisopropylethylamine, 2-(7-benzotriazole)-N,N,N’,N’- Two kinds of tetramethylurea hexafluorophosphate mixed in a 1:1 volume ratio, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc- Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 75°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated and washed twice with methyl tert-butyl ether, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 2:3 solution A-sodium phosphate buffered aqueous solution (concentration 15g/L), solution B-linear peptide atosiban crude peptide phosphoric acid aqueous solution and solution C: DMSO aqueous solution (volume 1:1).
[0074]
The crude peptide yield was 80%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70 DEG C for 28 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 55%.
[0075]
Example 4:
[0076]
Using Rink-Fmoc-Gly resin (40 g, substitution amount 0.36 mmol/g) as the starting material, the stepwise Fmoc-SPPS (solid phase peptide synthesis) method was used to synthesize the peptide. Fmoc deprotection was performed with piperidine in DMF (1:3 by volume). Subsequently, the other amino acids in the sequence were connected in the following order, and the coupling reagents were selected from 2-oxime ethyl cyanoacetate, N,N-diisopropylcarbodiimide, and 1-hydroxybenzotriazole in a volume ratio of 1. :1:1 mix, Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH , Fmoc-D-Tyr(ET)-OH, Mpa(Trt). Coupling and deprotection of amino acids were carried out at 80°C for 2-3 min and monitored with the Kaiser test. The peptide was cleaved with the lysing solution of TFA for 3 hours, precipitated with methyl tert-butyl ether and washed twice, and after centrifugal drying, the atosiban linear crude peptide was cyclized by the method of liquid phase synthesis, and the volume ratio was 1: 3:4 solution of A-trifluoroacetic acid-aqueous ammonia solution (concentration of 45 g/L), solution B-aqueous solution of linear peptide atosiban crude peptide trifluoroacetic acid and solution C: saturated aqueous iodine solution.
[0077]
The crude peptide yield was 78%. Crude atosiban was purified by preparative high performance liquid chromatography with a water/acetonitrile gradient from 100% water to 100% acetonitrile in 20 minutes. The purified atosiban solution is freeze-dried at -50 to -70° C. for 38 hours with a freeze dryer, the obtained atosiban has a purity of more than 99.5%, and the total product yield is 52%.
Atosiban Acetate Injection was first listed in Austria on March 23, 2000 under the trade name: Atosiban, a new type of anti-prematurity drug developed by Ferring GmbH, which is an oxytocin The analog is a competitive antagonist of oxytocin receptors in the uterus, decidua, and fetal membranes. It is a first-line drug recommended by the European Medical Association; it can inhibit the binding of oxytocin and oxytocin receptors, thereby directly inhibiting the effect of oxytocin. In the uterus, it can inhibit uterine contraction; it can also inhibit the hydrolysis of phosphatidylinositol.
Atosiban is a cyclic nonapeptide whose molecular formula is C 43 H 67 N 11 O 12 S 2 ; molecular weight is 994.19; CAS registration number is 90779-69-4; its peptide sequence is as follows:
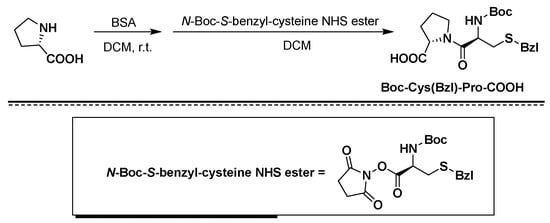
In the Chinese patents with announcement numbers CN101314613B and CN101696236B, the solid-phase synthesis of atosiban uses Rink Amide AM Resin resin solid-phase coupling stepwise to obtain Mpa(Trt)-D-Tyr(Et)-Ile-Thr(tBu)- Asn(Trt)-Cys(Trt)-Pro-Orn(Boc)-Gly-Resin is directly oxidized in solid phase to generate disulfide bonds, and then cleaved to obtain atosiban. The Rink Amide AM Resin resin used in the prior art needs to be cracked under a strong acid environment, which is not conducive to product stability and has a greater operational risk; Mpr and Cys both have sulfhydryl groups, and the sulfhydryl groups have the ability to capture tBu to generate double tBu impurities, When the peptide resin after solid-phase oxidation is cleaved to remove the protective group and resin, due to the presence of tBu or tBu source Boc protective group, it requires high capture agent, which is not conducive to product quality control and reduces product yield.
The Chinese patent with publication number CN105408344B discloses a method for synthesizing atosiban starting from Fmoc-Orn-Gly-NH2, wherein Fmoc-Orn-Gly-NH2 is connected to trityl through the side chain of ornithine On the base resin, impurities can be effectively controlled. However, using dipeptide and trityl-type resin for coupling, the resin attached to the Orn side chain of the dipeptide increases the steric hindrance of the subsequent Pro coupling and prolongs the coupling time, which is easy to cause missing peptide impurities.
Example 1. Synthesis of Fmoc-Pro-Orn-Gly-NH 2 tripeptide
[0027]
Fmoc-Pro-OH (134.94 g, 400 mmol) and N-hydroxysuccinimide (46.00 g, 400 mmol) were weighed into 1600 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (90.72g, 440mmol) in tetrahydrofuran (320ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 400 ml of tetrahydrofuran, and H-Orn(Boc)-NH 2 (92.92 g, 400 mmol) was dissolved in 300 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate to dryness under reduced pressure, add N-hydroxysuccinimide (46.00 g, 400 mmol) and 1600 ml of tetrahydrofuran to dissolve, and stir at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (90.72g, 440mmol) in tetrahydrofuran (320ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 400 ml of tetrahydrofuran, and H-Gly-NH 2 (29.64 g, 400 mmol) was dissolved in 300 ml of tetrahydrofuran and slowly added dropwise to the above solution, and the reaction was continued at room temperature after dropping, and the monitoring of the raw materials was completed. The reaction was filtered, and the filtrate was concentrated under reduced pressure. Dry, add 1000 mL of 5% TFA/DCM solution to the reaction solution, continue to react for 1 h, and concentrate to dryness to obtain a yellow oil, which is recrystallized from isopropanol to obtain 171.56 g of white solid with a yield of 69%.
[0028]
Example 2. Synthesis of Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.42 mmol/g
[0029]
Trityl resin (37.5 g, 30 mmol, substitution degree: 0.80 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 52.14 g of Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.42 mmol/g.
[0030]
Example 3. Synthesis of Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0031]
2-CTC Resin resin (30.0 g, 30 mmol, substitution degree: 1.00 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.80 g of Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0032]
Example 4. Synthesis of Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0033]
4-methyl-trityl resin (33.33 g, 30 mmol, substitution degree: 0.90 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.89 g of Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0034]
Example 5. Synthesis of Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 peptide resin with a degree of substitution of 0.50 mmol/g
[0035]
4-Methoxy-trityl resin (30.0 g, 30 mmol, substitution degree: 1.00 mmol/g) was weighed into a solid-phase reaction synthesis column. 400 mL of dry DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*400 mL of dry DMF, and the DMF was removed. Fmoc-Pro-Orn-Gly-NH 2 (37.30 g, 60 mmol) prepared in Example 1 , DIEA (11.63 g, 90 mmol) were added, 100 mL of dry DMF was added to dissolve and clarified, added to the resin to react for 2 h, and methanol (9.61 mmol) was added. g, 300 mmol) reacted for 20 min, sucked dry, washed the resin with 3*400 mL of CH 2 Cl 2 , and removed CH 2 Cl 2 . The resin was taken out and dried under vacuum at 25-35° C. to obtain 43.69 g of Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 resin with a measured substitution degree of 0.50 mmol/g.
[0036]
Example 6. Synthesis of Atosiban Linear Peptide Resin 1
[0037]
Fmoc-Pro-Orn (trityl resin)-Gly-NH 2 (35.71 g) prepared in Example 2 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0038]
Weigh 17.57g Fmoc-Cys(Trt)-OH and 4.86g HOBt, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.68g DIC (pre-cooled to <0°C), Activated in the solution for about 3 to 5 minutes, the activated solution was added to the reaction column under control, and reacted at 20 to 35 °C for 2 to 3 hours. The ninhydrin test was negative. The reaction solution was removed, and 200 mL of DMF was added to wash the resin. 6 times. After washing, the washing liquid was removed to obtain Fmoc-Cys(Trt)-Pro-Orn (trityl resin)-Gly-NH 2 .
[0039]
Repeat the step of receiving the peptide and remove the Fmoc protective group. According to the amino acid sequence of atosiban, Fmoc-Cys(Trt)-Pro-Orn (trityl resin)-Gly-NH 2 was coupled to Fmoc- Asn-OH, Fmoc-Thr-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(Et)-OH, Mpa(Trt)-OH give Mpa(Trt)-D-Tyr(Et)-Ile-Thr- Asn-Cys(Trt)-Pro-Orn (trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 48.72g after drying, and the resin weight gain was 89.0%.
[0040]
Example 7. Synthesis of atosiban linear peptide resin 2
[0041]
Fmoc-Pro-Orn(2-CTC Resin)-Gly-NH 2 (30.00 g) prepared in Example 3 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0042]
Weigh 17.57g Fmoc-Cys(Trt)-OH and 13.65g HBTU, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cooled to <0°C), put Activated in the solution for about 3 to 5 minutes, the activated solution was added to the reaction column under control, and reacted at 20 to 35 °C for 2 to 3 hours. The ninhydrin test was negative. The reaction solution was removed, and 200 mL of DMF was added to wash the resin. 6 times. After washing, the washing solution was removed to obtain Fmoc-Cys(Trt)-Pro-Orn(2-CTC Resin)-Gly-NH 2 .
[0043]
Fmoc-D-Tyr(Et)-OH (86.30 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and H-Ile-OH (26.24 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. The monitoring of the raw materials was completed. After filtration, the solution was concentrated under reduced pressure. , the concentrated solution was added to petroleum ether to separate out the solid, the solid was washed and then dried, recrystallized and dried with isopropanol to obtain 75.60 g of Fmoc-D-Tyr(Et)-Ile-OH with a yield of 75%.
[0044]
Repeat the step of receiving the peptide and removing the Fmoc protective group. According to the amino acid sequence of atosiban, sequentially couple Fmoc-Asn on Fmoc-Cys(Trt)-Pro-Orn(2-CTC Resin)-Gly-NH 2 -OH, Fmoc-Thr-OH, Fmoc-D-Tyr(Et)-Ile-OH, Mpa(Trt)-OH to give Mpa(Trt)-D-Tyr(Et)-Ile-Thr-Asn-Cys(Trt )-Pro-Orn( 2 -CTC Resin)-Gly-NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.77g after drying, and the resin weight gain rate was 87.4%.
[0045]
Example 8. Synthesis of atosiban linear peptide resin 3
[0046]
Fmoc-Pro-Orn (4-methyl-trityl resin)-Gly-NH 2 (30.00 g) prepared in Example 4 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod, and the ninhydrin test was positive, indicating that Fmoc had been removed.
[0047]
Weigh 17.57g Fmoc-Cys(Trt)-OH, 13.65g HBTU and 4.05g HOBt, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cooled to <0 ℃), activate in the solution for about 3-5min, add the activated solution to the reaction column, react at 20-35 ℃ for 2-3h, the ninhydrin test is negative, remove the reaction solution, add 200mL of DMF The resin was washed 6 times. After washing, the washing liquid was removed to obtain Fmoc-Cys(Trt)-Pro-Orn(4-methyl-trityl resin)-Gly-NH 2 .
[0048]
Mpa(Trt)-OH (69.69 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and HD-Tyr(Et)-OH (41.85 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate under reduced pressure, add the concentrated solution to petroleum ether to precipitate a solid, wash the solid and then dry it, recrystallize and dry with isopropanol to obtain Mpa(Trt)-D-Tyr(Et)-OH 77.98g, yield 72%.
[0049]
Repeat the step of receiving the peptide and removing the Fmoc protective group, according to the amino acid sequence of atosiban, on Fmoc-Cys(Trt)-Pro-Orn (4-methyl-trityl resin)-Gly- NH 2 Fmoc-Asn-OH, Fmoc-Thr-OH, Fmoc-Ile-OH, Mpa(Trt)-D-Tyr(Et)-OH were sequentially coupled to obtain Mpa(Trt)-D-Tyr(Et)-Ile-Thr -Asn-Cys(Trt)-Pro-Orn(4-methyl-trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.91g after drying, and the resin weight gain rate was 88.3%.
[0050]
Example 9. Synthesis of atosiban linear peptide resin 4
[0051]
Fmoc-Pro-Orn (4-methoxy-trityl resin)-Gly-NH 2 (30.00 g) prepared in Example 5 was weighed into a solid-phase reaction synthesis column. 400 mL of DMF was added to swell for 30 min, and the DMF was removed. The resin was washed with 3*200 mL of dry DMF, and the DMF was removed. 200 mL of DBLK solution (20% piperidine/DMF solution, V/V) was added and deprotected twice, the first time was 5 min and the second time was 15 min. After deprotection, the resin was washed with 200 mL of DMF each time, and washed 6 times. After the fourth washing, a little resin was taken with a glass rod. The ninhydrin test was positive, indicating that Fmoc had been removed.
[0052]
Fmoc-Asn-OH (70.87 g, 200 mmol) and N-hydroxysuccinimide (23.00 g, 200 mmol) were weighed into 800 ml of tetrahydrofuran, and stirred at room temperature. The temperature was controlled at about 5°C, and a solution of DCC (45.36g, 220mmol) in tetrahydrofuran (160ml) was slowly added and stirred at room temperature for 2.5h, filtered, concentrated and added to petroleum ether for recrystallization to precipitate a solid, washed and dried, and the obtained activated ester was The solid was dissolved in 200 ml of tetrahydrofuran, and H-Cys(Trt)-OH (79.96 g, 200 mmol) was dissolved in 150 ml of tetrahydrofuran and slowly added dropwise to the above solution. After dropping, the reaction was continued at room temperature. Concentrate under reduced pressure, add the concentrated solution to petroleum ether to precipitate a solid, wash the solid and then dry, recrystallize and dry with isopropanol to obtain Fmoc-Asn-Cys(Trt)-OH 102.17g, yield 73%.
[0053]
Weigh 20.99g Fmoc-Asn-Cys(Trt)-OH and 13.65g HCTU, add 100mL DMF to dissolve, after complete dissolution, cool the solution to below 5°C, then add 5.82g DIEA (pre-cool to <0°C) , activate in the solution for about 3-5min, add the activated solution to the reaction column, react at 20-35°C for 2-3h, the ninhydrin test is negative, remove the reaction solution, add 200mL of DMF to wash the resin , wash 6 times. After washing, the washing liquid was removed to obtain Fmoc-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly-NH 2 .
[0054]
Repeat the step of receiving the peptide and removing the Fmoc protective group. According to the amino acid sequence of atosiban, in Fmoc-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly- Fmoc-Thr-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(Et)-OH, Mpa(Trt)-OH were sequentially coupled on NH 2 to obtain Mpa(Trt)-D-Tyr(Et)-Ile- Thr-Asn-Cys(Trt)-Pro-Orn(4-methoxy-trityl resin)-Gly- NH2 . After washing with DMF, the washing solution was removed. The resin was washed with 200 ml of DCM each time, 4 times, 5 min/time, the DCM was removed, and the resin was vacuum-dried at room temperature (20-35° C.) until it was quicksand. The peptide resin was 42.28g after drying, and the resin weight gain was 84.0%.
[0055]
Example 10. Synthesis of atosiban crude peptide 1
[0056]
Configure 487.2ml of TFA/DCM=2/98 (V/V) lysis solution, cool to 5-10°C, add 48.72g of peptide resin prepared in Example 6 into the lysis solution, at room temperature (20-35°C) React for 5h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.77g of atosiban linear peptide, dissolve 14.30g of atosiban linear peptide in 0.75L of glacial acetic acid, add 6.75L of water to dilute, add 0.1M/L iodine ethanol solution dropwise until the solution changes color, react at room temperature for 1.0h, That is, the crude atosiban peptide is obtained, and its HPLC spectrum is shown in Figure 1.
[0057]
Example 11. Synthesis of atosiban crude peptide 2
[0058]
Configure TFA/DCM=5/95 (V/V) lysate 448.6ml, cool to 5~10℃, add 42.77g of peptide resin prepared in Example 7 into the lysate, at room temperature (20~35℃) React for 3h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate, dry to obtain a solid, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.21g of atosiban linear peptide, dissolve 14.21g of atosiban linear peptide in 1.5L of glacial acetic acid, add 6L of water to dilute, add 0.1M/L iodoethanol solution dropwise until the solution changes color, react at room temperature for 1.0h, that is The crude atosiban peptide was obtained, and its HPLC chromatogram was similar to that in Figure 1.
[0059]
Example 12. Synthesis of atosiban crude peptide 3
[0060]
Configure 450.5ml of TFA/DCM=20/80(V/V) lysis solution, cool to 5~10℃, add 45.05g of peptide resin prepared in Example 8 into the lysis solution, at room temperature (20~35℃) React for 2h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.63g of atosiban linear peptide, dissolve 14.63g of atosiban linear peptide in 1.5L of glacial acetic acid, add 6L of water to dilute, add 10% hydrogen peroxide solution, and react at room temperature for 1.0h to obtain atosiban Crude peptide, its HPLC chromatogram is similar to Figure 1.
[0061]
Example 13. Synthesis of atosiban crude peptide 4
[0062]
Configure TFA/DCM=1/99 (V/V) lysate 442.7ml, cool to 5~10℃, add 44.27g of peptide resin prepared in Example 9 into the lysate, at room temperature (20~35℃) React for 5h, filter, wash the peptide resin twice with acetonitrile, 50ml/time, combine into the filtrate, spin the filtrate to dry, obtain a solid after drying, wash with isopropyl ether, filter, and dry under reduced pressure at 20-35°C to constant weight To obtain 14.13 g of atosiban linear peptide, dissolve 14.13 g of atosiban linear peptide in 1.5 L of glacial acetic acid, add 6 L of water to dilute, add 30% hydrogen peroxide solution, and react at room temperature for 1.0 h to obtain atosiban Crude peptide, its HPLC chromatogram is similar to Figure 1.
[0063]
Example 14. Purification of atosiban crude peptide 1
[0064]
The atosiban crude peptide prepared in Example 10 was dissolved in 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.12g , the yield is 64%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is shown in Figure 2.
[0065]
Example 15. Purification of atosiban crude peptide 2
[0066]
The crude atosiban peptide obtained in Example 11 was dissolved in a 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 9.80 g , the yield is 62%, the purity is 99%, and the obtained atosiban peptide HPLC spectrum is similar to Figure 2.
[0067]
Example 16. Purification of atosiban crude peptide 3
[0068]
The crude atosiban peptide obtained in Example 12 was dissolved in a 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.28g , the yield is 65%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is similar to that in Figure 2.
[0069]
Example 17. Purification of atosiban crude peptide 4
[0070]
The crude atosiban peptide obtained in Example 13 was dissolved in 15% acetonitrile aqueous solution and filtered, purified by preparative reverse-phase HPLC (C18 column), transferred to salt, collected more than 99% of the fraction, concentrated and lyophilized to obtain 10.27g , the yield is 65%, the purity is 99%, and the HPLC spectrum of the obtained atosiban peptide is similar to that in Figure 2.
Atosiban is a nonapeptide which contains three non-natural amino acids: D-Tyr(Et), Mpa and Orn, and a pair of disulfide bonds looped between Mpa and Cys, the structural formula is: c[Mpa-D-Tyr(Et)-Ile-Thr-Asn-Cys]-Pro-Orn-Gly-NH2.
By means of competing for oxytocin receptor with oxytocin, Atosiban can inhibit the combination between oxytocin and oxytocin receptor, and directly prevent the oxytocin from acting on uterus, and then inhibit the uterine contraction; as another hand, atosiban can also inhibit the hydrolysis of phosphatidylinositol and then block the generation of messenger and activity of Ca2+, with the decreasing of activity from oxytocin, the contraction of uterine is indirectly inhabited.
At present, there are many reports about synthesis process method in China and abroad A report in China shows that the inventor found a simple process by adopting solid phase oxidation, resulting in a low purity crude product, with low yield and low application value. The aforementioned reports about atosiban synthesis process reveal that most of them adopt the method using Boc solid phase synthetic and cleaving peptide with liquid ammonia, then oxidating with liquid phase oxidation, and purifying. Those respective processes result in “the three wastes” and are too complex for industrial production. See U.S. Pat. No. 4,504,469.
Example 1Preparing the Linear Atosiban Peptide Resin
(i) 6.25 g of Rink Amide resin (substitutability=0.8 mmol/g) is put into a reaction bottle, DMF is added into the bottle and washed twice, then swelled for 30 min with DMF. Fmoc protecting group of Rink Amide resin is removed with 30-40 ml of 20% DBLK, washed for 4 times with DMF, then washed twice with DCM after removal, the product is detected by ninhydrin detecting method, the resin is reddish-brown.
(ii) 4.46 g of Fmoc-Gly-OH and 2.43 g of HOBt dissolved in a suitable amount of DMF, which had been pre-activated with 3.05 ml DIC; the mixture is, added to the reaction bottle, and reacted for 2 h, the resin is negative by ninhydrin detecting method, after the reaction, the product is washed for 4 times with DMF, then washed twice with DCM, if the resin is positive, repeating the above condensation reaction until negative.
(iii) Fmoc-Orn(Boc)-OH, Fmoc-Pro-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Ile-OH, Fmoc-D-Tyr(ET)-OH and Mpa(Trt)-OH are coupled orderly.
Example 2Cleaving the Linear Atosiban Peptide Resin
5.15 g of linear atosiban is prepared by washing the linear atosiban peptide resin obtained from Example 1 for 3 times with 30 ml of methanol, adding the dry resin obtained to 150 ml of mixed solution with a volume ratio of TFA:H2O=95:5, reacting for 2 hours at 25° C. and filtering, washing the resin for 3 times with few trifluoroacetic acid, combining the filtrate and pouring into 1500 ml glacial ether, making rest for 2 hours, centrifugally separating the linear atosiban, washing for 3 times, and drying in a vacuum drier, MS: 995.3, HPLC: 91.5%, content: 65.5%, synthesis yield: 68%.
Example 3Oxidizing the Linear Atosiban
2.85 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 5% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.60 g of H2O2, reacting for 10 min at 25° C., monitoring with HPLC (HPLC: 75.6%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.4%.
Example 4Oxidizing the Linear Atosiban
3.01 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 10% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.85 g of H2O2, reacting for 30 min at 25° C., monitoring with HPLC (HPLC: 89.5%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.6%.
Example 5Oxidizing the Linear Atosiban
2.95 g of atosiban acetate is prepared by dissolving the linear atosiban obtained from Example 2 in 250 ml of 10% acetonitrile aqueous solution, adjusting the pH value to 8 to 9 with 30% ammonia water, adding 0.85 g of H2O2, reacting for 60 min at 25° C., monitoring with HPLC (HPLC: 83.5%), filtering after reaction, purifying filtrate by preparative RP-HPLC (column C18 or C8), transferring salt, and freeze-drying, MS: 994.5, HPLC: 99.4%.
The above is the further detailed description of the invention in conjunction with specific preferred examples, but it should not be considered that the specific examples of the invention are only limited to the these descriptions. For one of ordinary skill in the art, many deductions and replacements can be made without departing from the inventive concept. Such deductions and replacements should fall within the scope of protection of the invention.
A 2013 retrospective study comparing the efficacy and safety of atosiban and nifedipine in the suppression of preterm labour concluded that atosiban and nifedipine are effective in delaying delivery for seven days or more in women presenting with preterm labour.[16] A total of 68.3% of women in the atosiban group remained undelivered at seven days or more, compared with 64.7% in the nifedipine group.[16] They have the same efficacy and associated minor side effects.[16] However, flushing, palpitation, and hypotension were significantly higher in the nifedipine group.[16]
A 2012 clinical trial compared tocolytic efficacy and tolerability of atosiban with that of nifedipine.[17] Forty-eight (68.6%) women allocated to atosiban and 39 (52%) to nifedipine did not deliver and did not require an alternate agent at 48 hours, respectively (p=.03).[17] Atosiban has fewer failures within 48 hours.[17] Nifedipine may be associated with a longer postponement of delivery.[17]
A 2009 randomised controlled study demonstrated for the first time the direct effects of atosiban on fetal movement, heart rate, and blood flow.[18] Tocolysis with either atosiban or nifedipine combined with betamethasone administration had no direct fetal adverse effects.[18]
Atosiban vs. ritodrine
Multicentre, controlled trial of atosiban vs. ritodrine in 128 women shows a significantly better tocolytic efficacy after 7 days in the atosiban group than in the ritodrine group (60.3 versus 34.9%), but not at 48 hours (68.3 versus 58.7%). Maternal adverse events were reported less frequently in the atosiban group (7.9 vs 70.8%), resulting in fewer early drug terminations due to adverse events (0 versus 20%). Therefore, atosiban is superior to ritodrine in the treatment of preterm labour.[19]
Brand names
In India it is marketed under the brand name Tosiban by Zuventus healthcare ltd.
^ Akerlund M, Carlsson AM, Melin P, Trojnar J (1985). “The effect on the human uterus of two newly developed competitive inhibitors of oxytocin and vasopressin”. Acta Obstet Gynecol Scand. 64 (6): 499–504. doi:10.3109/00016348509156728. PMID4061066. S2CID25799128.
^ Lamont, Ronald F; Kam, KY Ronald (March 2008). “Atosiban as a tocolytic for the treatment of spontaneous preterm labor”. Expert Review of Obstetrics & Gynecology. 3 (2): 163–174. doi:10.1586/17474108.3.2.163. ISSN1747-4108.
^ Jump up to:abc Lamont, Ronald F.; Kam, KY Ronald (2008). “Atosiban as a tocolytic for the treatment of spontaneous preterm labor”. Expert Review of Obstetrics & Gynecology. 3 (2): 163–174. doi:10.1586/17474108.3.2.163.
^ Lamont CD, Jørgensen JS, Lamont RF (September 2016). “The safety of tocolytics used for the inhibition of preterm labour”. Expert Opinion on Drug Safety. 15 (9): 1163–73. doi:10.1080/14740338.2016.1187128. PMID27159501. S2CID4937942. It was for this reason and the fact that Tractocile (atosiban) only had a short duration before it was out of patent that the parent drug company decided not to pursue licensing in the USA.
^ Coomarasamy, A; Knox, EM; Gee, H; Khan, KS (November 2002). “Oxytocin antagonists for tocolysis in preterm labour — a systematic review”. Med Sci Monit. 8 (11): RA268–73. PMID12444392.
^ Jump up to:ab de Heus R, Mulder EJ, Derks JB, Visser GH (June 2009). “The effects of the tocolytics atosiban and nifedipine on fetal movements, heart rate and blood flow”. J Matern Fetal Neonatal Med. 22 (6): 485–90. doi:10.1080/14767050802702349. PMID19479644. S2CID35810758.
^ Shim JY, Park YW, YoonBH, Cho YK, Yang JH, Lee Y, Kim A. “Multicentre, parallelgroup, randomised, single-blind study of the safety and efficacy of atosibanversus ritodrine in the treatment of acute preterm labour in Korean women. BJOG 2006Nov;113(11):1228-34.
External links
“Atosiban”. Drug Information Portal. U.S. National Library of Medicine.
The most common adverse reactions include reduced platelet and other blood cell levels, as well as mucositis, febrile neutropenia, vomiting, pyrexia (fever), alopecia (hair loss), epistaxis (nosebleed), abdominal pain, musculoskeletal pain, cough, headache, diarrhea, rash, constipation, nausea, decreased appetite, pigmentation disorder and pruritus (itch).[5]
It was approved for medical use in the European Union in May 2019,[2] and in the United States in August 2022.[5]
FDA Approves First Cell-Based Gene Therapy to Treat Adult and Pediatric Patients with Beta-thalassemia Who Require Regular Blood Transfusions
Today, the U.S. Food and Drug Administration approved Zynteglo (betibeglogene autotemcel), the first cell-based gene therapy for the treatment of adult and pediatric patients with beta-thalassemia who require regular red blood cell transfusions.
“Today’s approval is an important advance in the treatment of beta-thalassemia, particularly in individuals who require ongoing red blood cell transfusions,” said Peter Marks, M.D., Ph.D., director of the FDA’s Center for Biologics Evaluation and Research. “Given the potential health complications associated with this serious disease, this action highlights the FDA’s continued commitment to supporting development of innovative therapies for patients who have limited treatment options.”
Beta-thalassemia is a type of inherited blood disorder that causes a reduction of normal hemoglobin and red blood cells in the blood, through mutations in the beta-globin subunit, leading to insufficient delivery of oxygen in the body. The reduced levels of red blood cells can lead to a number of health issues including dizziness, weakness, fatigue, bone abnormalities and more serious complications. Transfusion-dependent beta-thalassemia, the most severe form of the condition, generally requires life-long red blood cell transfusions as the standard course of treatment. These regular transfusions can be associated with multiple health complications of their own, including problems in the heart, liver and other organs due to an excessive build-up of iron in the body.
Zynteglo is a one-time gene therapy product administered as a single dose. Each dose of Zynteglo is a customized treatment created using the patient’s own cells (bone marrow stem cells) that are genetically modified to produce functional beta-globin (a hemoglobin component).
The safety and effectiveness of Zynteglo were established in two multicenter clinical studies that included adult and pediatric patients with beta-thalassemia requiring regular transfusions. Effectiveness was established based on achievement of transfusion independence, which is attained when the patient maintains a pre-determined level of hemoglobin without needing any red blood cell transfusions for at least 12 months. Of 41 patients receiving Zynteglo, 89% achieved transfusion independence.
The most common adverse reactions associated with Zynteglo included reduced platelet and other blood cell levels, as well as mucositis, febrile neutropenia, vomiting, pyrexia (fever), alopecia (hair loss), epistaxis (nosebleed), abdominal pain, musculoskeletal pain, cough, headache, diarrhea, rash, constipation, nausea, decreased appetite, pigmentation disorder and pruritus (itch).
There is a potential risk of blood cancer associated with this treatment; however, no cases have been seen in studies of Zynteglo. Patients who receive Zynteglo should have their blood monitored for at least 15 years for any evidence of cancer. Patients should also be monitored for hypersensitivity reactions during Zynteglo administration and should be monitored for thrombocytopenia and bleeding.
Betibeglogene autotemcel is made individually for each recipient out of stem cells collected from their blood, and must only be given to the recipient for whom it is made.[2] It is given as an autologous intravenous infusion and the dose depends on the recipient’s body weight.[3][2]
To make betibeglogene autotemcel, the stem cells taken from the recipient’s blood are modified by a virus that carries working copies of the beta globin gene into the cells.[2] When these modified cells are given back to the recipient, they are transported in the bloodstream to the bone marrow where they start to make healthy red blood cells that produce beta globin.[2] The effects of betibeglogene autotemcel are expected to last for the recipient’s lifetime.[2]
Mechanism of action
Beta thalassemia is caused by mutations to or deletions of the HBB gene leading to reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals.[8] LentiGlobin BB305 is a lentiviral vector which inserts a functioning version of the HBB gene into a recipient’s blood-producing hematopoietic stem cells (HSC) ex vivo. The resulting engineered HSCs are then reintroduced to the recipient.[9][10]
History
In early clinical trials several participants with beta thalassemia, who usually require frequent blood transfusions to treat their disease, were able to forgo blood transfusions for extended periods of time.[11][12][13] In 2018, results from phase 1-2 trials suggested that of 22 participants receiving Lentiglobin gene therapy, 15 were able to stop or reduce regular blood transfusions.[14][15]
The safety and effectiveness of betibeglogene autotemcel were established in two multicenter clinical studies that included adult and pediatric particpiants with beta-thalassemia requiring regular transfusions.[5] Effectiveness was established based on achievement of transfusion independence, which is attained when the particpiant maintains a pre-determined level of hemoglobin without needing any red blood cell transfusions for at least 12 months. Of 41 particpiants receiving betibeglogene autotemcel, 89% achieved transfusion independence.[5]
Society and culture
Legal status
It was approved for medical use in the European Union in May 2019,[2] and in the United States in August 2022.[5]
^ Jump up to:ab Clinical trial number NCT02140554 for “A Phase 1/2 Study Evaluating Gene Therapy by Transplantation of Autologous CD34+ Stem Cells Transduced Ex Vivo With the LentiGlobin BB305 Lentiviral Vector in Subjects With Severe Sickle Cell Disease” at ClinicalTrials.gov
Tolebrutinib (R&D code SAR442168), developed by Principia and later acquired by Sanofi and included in its product line, Tolebrutinib is a BTK inhibitor used to treat cancer, autoimmune diseases such as multiple sclerosis and myasthenia gravis, inflammatory diseases and thromboembolic diseases, etc.,
Tolebrutinib is an orally bioavailable, brain-penetrant, selective, small molecule inhibitor of Bruton’s tyrosine kinase (BTK), with potential immunomodulatory and anti-inflammatory activities. Upon oral administration, tolebrutinib is able to cross the blood-brain barrier and inhibits the activity of BTK both peripherally and in the central nervous system (CNS). This prevents the activation of the B-cell antigen receptor (BCR) signaling pathway, and the resulting immune activation and inflammation. The inhibition of BTK activity also prevents microglial inflammatory signaling in the CNS, and the resulting immune activation, neuroinflammation and neurodegeneration. BTK, a cytoplasmic tyrosine kinase and member of the Tec family of kinases, plays an important role in B lymphocyte development, activation, signaling, proliferation and survival. In addition to B cells, BTK is also expressed in innate immune cells, including macrophages and microglia, and plays an important role in the regulation of microglial inflammatory signaling.
BTK, a member of the Tec family non-receptor tyrosine kinases, is essential for B cell signaling downstream from the B-cell receptor. It is expressed in B cells and other hematopoietic cells such as monocytes, macrophages and mast cells. It functions in various aspects of B cell function that maintain the B cell repertoire (see Gauld S. B. et al., B cell antigen receptor signaling: roles in cell development and disease. Science,
296: 1641 -2. 2002.) B cells pay a role in rheumatoid arthritis (see Perosa F., et ai, CD20-depleting therapy in autoimmune diseases: from basic research to the clinic. / Intern Med. 267:260-77. 2010 and Dorner T, et at. Targeting B cells in immune-mediated
inflammatory disease: a comprehensive review of mechanisms of action and identification of biomarkers. Pharmacol The 125:464-75. 2010 and Honigberg, L., et. ai, The selective BTK inhibitor PCI-32765 blocks B cell and mast cell activation and prevents mouse collagen indiced arthritis. Clin. Immunol. 127 SI :S 111. 2008) and in other autoimmune diseases such as systemic lupus erythematosus and cancers (see Shlomchik M. J., et. ai, The role of B cells in lpr/lpr-induced autoimmunity. /. Exp Med. 180:1295-1306. 1994; Honigberg L. A., The Braton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. 107: 13075-80. 2010; and Mina-Osorio P, et al., Suppression of
glomerulonephritis in lupus-prone NZB x NZW mice by RN486, a selective inhibitor of Bruton’s tyrosine kinase. Arthritis Rheum. 65: 2380-91. 2013).
There is also potential for BTK inhibitors for treating allergic diseases (see Honigberg, L., et. al., The selective BTK inhibitor PCI-32765 blocks B cell and mast cell activation and prevents mouse collagen indiced arthritis. Clin. Immunol. 127 SI :S111. 2008). It was noted that the irreversible inhibitor suppresses passive cutaneous anaphylaxis (PCA) induced by IgE antigen complex in mice. These findings are in agreement with those noted with BTK-mutant mast cells and knockout mice and suggest that BTK inhibitors may be useful for the treatment of asthma, an IgE-dependent allergic disease of the airway.
Accordingly, compounds that inhibit BTK would be useful in treatment for diseases such as autoimmune diseases, inflammatory diseases, and cancer.
Synthesis of (R)-l-(l-acryloylpiperidin-3-yl)-4-amino-3-(4-phenoxyphenyl)-lH- imidazo[4,5-c]pyridin-2(3H)-one
Into a 100-mL round-bottom flask, was placed (R)-4-amino-3-(4-phenoxyphenyl)-l-(piperidin-3-yl)-lH-imidazo[4,5-c]pyridin-2(3H)-one (150 mg, 0.37 mmol, 1.00 equiv), DCM-CH30H (6 mL), TEA (113 mg, 1.12 mmol, 3.00 equiv). This was followed by the addition of prop-2-enoyl chloride (40.1 mg, 0.44 mmol, 1.20 equiv) dropwise with stirring at OoC in 5 min. The resulting solution was stirred for 2 h at 0 °C. The resulting mixture was concentrated under vacuum. The residue was applied onto a silica gel column with dichloromethane/methanol (30: 1). The crude product (100 mg) was purified by Prep-HPLC with the following conditions (Column, XBridge Prep CI 8 OBD
Column,5um, 19*150mm; mobile phase, water with 0.05%TFA and ACN (25.0% ACN up to 45.0% in 8 min). 54.5 mg product of (R)-l-(l -acryloylpiperidin-3-yl)-4-amino-3-(4-phenoxyphenyl)-lH-imidazo[4,5-c]pyridin-2(3H)-one was obtained as a white solid. LC-MS m/z: 465.2 (M+l)
Step 2
Into a 25-mL round-bottom flask was placed tert-butyl (3R)-3-[4-[(E)-[(dimethy]amino)-methylidene]-amino]-2-oxo-3-(4-phenoxyphenyl)-lH,2H,3H-imidazo[4,5-c]pyridin-l -yl]piperidine- l-carboxylate (150 mg, 0.27 mmol, 1.00 equiv), 1,4-dioxane (6 mL), and hydrogen chloride (3 mL). The resulting solution was stirred overnight at 50° C. The reaction mixture was quenched with water. The pH of the solution was adjusted to 9 with sodium bicarbonate. The resulting solution was extracted with dichloromethane:CH3OH=10: 1 and the organic layers were combined. The resulting mixture was washed with sodium chloride and the organic layers were combined, dried over anhydrous sodium sulfate and concentrated under vacuum. The residue was applied onto a silica gel column and eluted with dichloromethane/methanol (30: 1) to give 80 mg (74%) of 4-amino-3-(4-phenoxyphenyl)-l -[(3R)-piperidin-3-yl]-lH,2H,3H-imidazo[4,5-c]pyridin-2-one as a light yellow solid.
////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The Bcl-2 family is most notable for their regulation of apoptosis, a form of programmed cell death, at the mitochondrion; Bcl-2 and Bcl-xL are anti-apoptotic proteins. Because many cancers have mutations in these genes that allow them to survive, scientists began working to develop drugs that would inhibit this pathway in the 1990s.[1] ABT-737 was one of the earliest of a series of drugs developed by Abbott Laboratories (now Abbvie) to target this pathway, based on their resolution of the 3D structure of Bcl-xL and studies using high-field solution nuclear magnetic resonance (NMR) that revealed how the BH domains of these proteins interacted with their targets.[1]
ABT-737 was superior to previous BCL-2 inhibitors given its higher affinity for Bcl-2, Bcl-xL and Bcl-w. In vitro studies showed that primary cells from patients with B-cell malignancies are sensitive to ABT-737.[3] In animal models, it improved survival, caused tumor regression, and cured a high percentage of mice.[4] In preclinical studies utilizing patient xenografts, ABT-737 showed efficacy for treating lymphoma and other blood cancers.[5]
Subsequently, it was reported that ABT-737 specifically induces apoptosis in senescent cells in vitro and in mouse models.[2]
ABT-737, a BH3 mimetic, is a potent Bcl-2, Bcl-xL and Bcl-w inhibitor with EC50s of 30.3 nM, 78.7 nM, and 197.8 nM, respectively. ABT-737 induces the disruption of the BCL-2/BAX complex and BAK-dependent but BIM-independent activation of the intrinsic apoptotic pathway. ABT-737 induces autophagy and has the potential for acute myeloid leukemia (AML) research.
^ Vogler, Meike, et al. “Bcl-2 inhibitors: small molecules with a big impact on cancer therapy.” Cell Death & Differentiation 16.3 (2008): 360–367.
^ Oltersdorf, Tilman; Elmore, Steven W.; Shoemaker, Alexander R.; Armstrong, Robert C.; Augeri, David J.; Belli, Barbara A.; Bruncko, Milan; Deckwerth, Thomas L.; Dinges, Jurgen; Hajduk, Philip J.; Joseph, Mary K.; Kitada, Shinichi; Korsmeyer, Stanley J.; Kunzer, Aaron R.; Letai, Anthony; Li, Chi; Mitten, Michael J.; Nettesheim, David G.; Ng, ShiChung; Nimmer, Paul M.; O’Connor, Jacqueline M.; Oleksijew, Anatol; Petros, Andrew M.; Reed, John C.; Shen, Wang; Tahir, Stephen K.; Thompson, Craig B.; Tomaselli, Kevin J.; Wang, Baole; Wendt, Michael D.; Zhang, Haichao; Fesik, Stephen W.; Rosenberg, Saul H. (2005). “An inhibitor of Bcl-2 family proteins induces regression of solid tumours”. Nature. 435 (7042): 677–81. Bibcode:2005Natur.435..677O. doi:10.1038/nature03579. PMID15902208. S2CID4335635.
Are you aware of any Chemical Database which offers one stop solution to the Sourcing, R&D and Business Development department? Explore Smartchem to Quickly find Suppliers (Procurement), Customers (BD) & Synthetic pathways (R&D)
Is this the information you looking for? Evaluate SmartChem, lets schedule a demo. Try us once. You will use us for life.
Zuranolone was approved for medical use in the United States for the treatment of postpartum depression in August 2023.[2] It was developed by Sage Therapeutics and Biogen.[9]
The most common side effects include drowsiness, dizziness, diarrhea, fatigue, nasopharyngitis (cold-like symptoms), and urinary tract infection.[2]
The US FDA label contains a boxed warning noting that zuranolone can impact a person’s ability to drive and perform other potentially hazardous activities.[2] Use of zuranolone may cause suicidal thoughts and behavior.[2] Zuranolone may cause fetal harm.[2]
History
Zuranolone was developed as an improvement on the intravenously administered neurosteroid brexanolone, with high oral bioavailability and a biological half-life suitable for once-daily administration.[7][10] Its half-life is around 16 to 23hours, compared to approximately 9hours for brexanolone.[4][5]
The efficacy of zuranolone for the treatment of postpartum depression in adults was demonstrated in two randomized, double-blind, placebo-controlled, multicenter studies.[2] The trial participants were women with postpartum depression who met the Diagnostic and Statistical Manual of Mental Disorders criteria for a major depressive episode and whose symptoms began in the third trimester or within four weeks of delivery.[2] In study 1, participants received 50 mg of zuranolone or placebo once daily in the evening for 14 days.[2] In study 2, participants received another zuranolone product that was approximately equal to 40 mg of zuranolone or placebo, also for 14 days.[2] Participants in both studies were monitored for at least four weeks after the 14-day treatment.[2] The primary endpoint of both studies was the change in depressive symptoms using the total score from the 17-item Hamilton depression rating scale (HAMD-17), measured at day 15.[2] Participants in the zuranolone groups showed significantly more improvement in their symptoms compared to those in the placebo groups.[2] The treatment effect was maintained at day 42—four weeks after the last dose of zuranolone.[2]
Zuranolone was approved by the US Food and Drug Administration (FDA) for the treatment of postpartum depression in August 2023.[2][12] The FDA granted the application for zuranolone priority review and fast track designations.[2] Approval of Zurzuvae was granted to Sage Therapeutics, Inc.[2]
Zuranolone has also been under development for the treatment of major depressive disorder, but the application for this use was given a Complete Response Letter (CRL) by the FDA due to insufficient evidence of effectiveness.[13]
Research
In a randomized, placebo-controlled phase III trial to assess its efficacy and safety for the treatment of major depressive disorder, subjects in the zuranolone group (50 mg oral zuranolone once daily for 14 days) experienced statistically significant and sustained improvements in depressive symptoms (as measured by HAM-D score) throughout the treatment and follow-up periods of the study.[14]
Example 1. Synthesis of 1-(2-((3R,5R,8R,9R,10S,13S,14S,17S)-3-hydroxy-3,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-17-yl)-2-oxoethyl)-1H-pyrazole-4-carbonitrile (Compound 1).
[00488] To a suspension of K2CO3 (50 mg, 0.36 mmol) in THF (5 mL) was added 1H-pyrazole-4-carbonitrile (100 mg, 0.97 mmol) and 2-bromo-1-((3R,5R,8R,9R,10S,13S,14S,17S)-3-hydroxy-3,13-dimethylhexadecahydro-1H-cyclopenta[ ^]phenanthren-17-yl)ethan-1-one (50 mg, 0.12 mmol). The mixture was stirred at room temperature for 15 hours. The reaction mixture was poured into 5 mL H2O and extracted with ethyl acetate (2×10 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue mixture was purified by reverse-phase preparative HPLC to afford Compound 1 as a white solid (9 mg, 17.4% yield).1H NMR (500 MHZ, CDCl3) δ (ppm) 7.87 (1H, s), 7.82 (1H, s), 5.02 (1H, AB), 4.2 (1H, AB), 2.61 (1H, t), 2.16-2.24 (1H, m), 2.05 (1H, dxt), 1.70-1.88 (6H, m), 1.61-1.69 (2H, m), 1.38-1.52 (6H, m), 1.23-1.38 (5H, m), 1.28 (3H, s), 1.06-1.17 (3H, m), 0.67 (3H, s). LCMS: rt=2.24 min, m/z=410.1 [M+H]+.
PAPER
Journal of Medicinal Chemistry (2017), 60(18), 7810-7819
Certain classes of neuroactive steroids (NASs) are positive allosteric modulators (PAM) of synaptic and extrasynaptic GABAA receptors. Herein, we report new SAR insights in a series of 5β-nor-19-pregnan-20-one analogues bearing substituted pyrazoles and triazoles at C-21, culminating in the discovery of 3α-hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217, 3), a potent GABAA receptor modulator at both synaptic and extrasynaptic receptor subtypes, with excellent oral DMPK properties. Compound 3 has completed a phase 1 single ascending dose (SAD) and multiple ascending dose (MAD) clinical trial and is currently being studied in parallel phase 2 clinical trials for the treatment of postpartum depression (PPD), major depressive disorder (MDD), and essential tremor (ET).
The compounds of the invention can be prepared in accordance with methods described in the art (Upasmi et al., J. Med. Chem. 1997, 40:73-84; and Hogenkamp et al., J. Med. Chem. 1997, 40:61- 72) and using the appropriate reagents, starting materials, and purification methods known to those skilled in the art. In some embodiments, compounds described herein can be prepared using methods shown in general Schemes 1-4, comprising a nucleophilic substitution of 19-nor pregnane bromide with a neucleophile. In certain embodiments, the nucleophile reacts with the 19-nor pregnane bromide in the presence of K2CO3 in THF.
Synthesis of compound SA-B. Compound SA (50 g, 184 mmol) and palladium black (2.5 g) in tetrahydrofuran (300 mL) and concentrated hydrobromic acid (1.0 mL) was hydrogenated with 10 atm hydrogen. After stirring at room temperature for 24h, the mixture was filtered through a pad of celite and the filtrate was concentrated in vacuo to afford the crude compound. Recrystallization from acetone gave compound SA-B (42.0 g, yield: 83.4%) as white powder.
Synthesis of compound SA-C. A solution of SA-B (42.0 g, 153.06 mmol) in 600 mL anhydrous toluene was added dropwise to the methyl aluminum bis(2,6-di-tert-butyl-4-methylphenoxide (MAD) (459.19 mmol, 3.0 eq, freshly prepared) solution under N2 at -78°C. After the addition was completed, the reaction mixture was stirred for 1 hr at -78°C. Then 3.0 M MeMgBr (153.06 mL, 459.19 mmol) was slowly added dropwise to the above mixture under N2 at -78°C. Then the reaction mixture was stirred for 3 hr at this temperature. TLC (Petroleum ether/ethyl acetate = 3:1) showed the reaction was completed. Then saturated aqueous NH4Cl was slowly added dropwise
to the above mixture at -78°C. After the addition was completed, the mixture was filtered, the filter cake was washed with EtOAc, the organic layer was washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated, purified by flash Chromatography on silica gel (Petroleum ether/ ethyl acetate20:1 to 3:1) to afford compound SA-C (40.2 g, yield: 90.4%) as white powder. 1H NMR: (400 MHz, CDCl3) δ 2.47-2.41 (m, 1H), 2.13-2.03 (m, 1H), 1.96-1.74 (m, 6H), 1.70-1.62 (m, 1H), 1.54-1.47 (m, 3H), 1.45-1.37 (m, 4H), 1.35-1.23 (m, 8H), 1.22-1.10 (m, 2H), 1.10-1.01 (m, 1H), 0.87 (s, 3H).
Synthesis of compound SA-D. To a solution of PPh3EtBr (204.52 g, 550.89 mmol) in THF (500 mL) was added a solution of t-BuOK (61.82 g, 550.89 mmol) in THF (300 mL) at 0°C. After the addition was completed, the reaction mixture was stirred for 1 h 60 °C, then SA-C (40.0 g, 137.72 mmol) dissolved in THF (300 mL) was added dropwise at 60°C. The reaction mixture was heated to 60 °C for 18 h. The reaction mixture was cooled to room temperature and quenched with Sat. NH4Cl, extracted with EtOAc (3*500 mL). The combined organic layers were washed with brine, dried and concentrated to give the crude product, which was purified by a flash column chromatography (Petroleum ether/ ethyl acetate50:1 to 10:1) to afford compound SA-D (38.4 g, yield:92%) as a white powder. 1H NMR: (400 MHz, CDCl3) δ 5.17-5.06 (m, 1H), 2.42-2.30 (m, 1H), 2.27-2.13 (m, 2H), 1.89-1.80 (m, 3H), 1.76-1.61 (m, 6H), 1.55-1.43 (m, 4H), 1.42-1.34 (m, 3H), 1.33-1.26 (m, 6H), 1.22-1.05 (m, 5H), 0.87 (s, 3H).
Synthesis of compound SA-E. To a solution of SA-D (38.0 g, 125.62 mmol) in dry THF (800 mL) was added dropwise a solution of BH3.Me2S (126 mL, 1.26 mol) under ice-bath. After the addition was completed, the reaction mixture was stirred for 3 h at room temperature (14-20 °C). TLC (Petroleum ether/ ethyl acetate3:1) showed the reaction was completed. The mixture was cooled to 0 °C and 3.0 M aqueous NaOH solution (400 mL) followed by 30% aqueous H2O2 (30%, 300 mL) was added. The mixture was stirred for 2 h at room temperature (14-20 °C), and then filtered, extracted with EtOAc (3*500 mL). The combined organic layers were washed with saturated aqueous Na2S2O3, brine, dried over Na2SO4 and concentrated in vacuum to give the crude product (43 g , crude) as colorless oil. The crude product was used in the next step without further purification.
Synthesis of compound SA-F. To a solution of SA-E (43.0 g, 134.16 mmol) in dichloromethane (800 mL) at 0 °C and PCC (53.8 g, 268.32 mmol) was added portion wise. Then the reaction mixture was stirred at room temperature (16-22 °C) for 3 h. TLC (Petroleum ether/ ethyl acetate3:1) showed the reaction was completed, then the reaction mixture was filtered, washed with DCM. The organic phase was washed with saturated aqueous Na2S2O3, brine, dried over Na2SO4 and concentrated in vacuum to give the crude product. The crude product was purified by a flash column chromatography (Petroleum ether/ ethyl acetate50:1 to 8:1) to afford compound SA-F (25.0 g, yield:62.5%, over two steps) as a white powder. 1H NMR (SA-F): (400 MHz, CDCl3) δ 2.57-2.50 (m, 1H), 2.19-2.11 (m, 4H), 2.03-1.97 (m, 1H), 1.89-1.80 (m, 3H), 1.76-1.58 (m, 5H), 1.47-1.42 (m, 3H), 1.35-1.19 (m, 10H), 1.13-1.04 (m, 3H), 0.88-0.84 (m, 1H), 0.61 (s, 3H).
Synthesis of compound SA. To a solution of SA-F (10 g, 31.4 mmol) and aq. HBr (5 drops, 48% in water) in 200 mL of MeOH was added dropwise bromine (5.52 g, 34.54 mmol). The reaction mixture was stirred at 17 °C for 1.5 h. The resulting solution was quenched with saturated aqueous NaHCO3 at 0°C and extracted with EtOAc (150 mLx2). The combined organic layers were dried and concentrated. The residue was purified by column chromatography on silica gel eluted with (PE: EA=15:1 to 6:1) to afford compound SA (9.5 g, yield: 76.14%) as a white solid. LC/MS: rt 5.4 mm ; m/z 379.0, 381.1, 396.1.
To a suspension of K2CO3 (50 mg, 0.36mmol) in THF (5 mL) was added ethyl 1H-pyrazole-4-carbonitrile (100 mg, 0.97 mmol ) and SA (50 mg,0.12 mmol). The mixture was stirred at rt for 15h. The reaction mixture was poured in to 5 mL H2O and extracted with EtOAc (2 x 10 mL). The combined organic layers were washed with brine, dried over sodium sulfate, filtered and concentrated. The residue mixture was purified with by reverse-phase prep-HPLC to afford the title compound as a white solid (9mg, 17.4%). 1H NMR (500 MHz, CDCl3), δ (ppm) 7.87 (1H, s),
7.82 (1H, s), 5.02 (1H, AB), 4.92 (1H, AB), 2.61 (1H, t), 2.16-2.24 (1H, m), 2.05 (1H, dXt), 1.70-1.88 (6H, m), 1.61-1.69 (2H, m), 1.38-1.52 (6H, m), 1.23-1.38 (5H, m), 1.28 (3H, s), 1.06-1.17 (3H, m), 0.67 (3H, s). LCMS: rt = 2.24 mm, m/z = 410.1 [M+H]+.
PATENT
WO2020150210
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
^ Jump up to:abBlanco MJ, La D, Coughlin Q, Newman CA, Griffin AM, Harrison BL, et al. (2018). “Breakthroughs in neuroactive steroid drug discovery”. Bioorganic & Medicinal Chemistry Letters. 28 (2): 61–70. doi:10.1016/j.bmcl.2017.11.043. PMID29223589.
^Martinez Botella G, Salituro FG, Harrison BL, Beresis RT, Bai Z, Blanco MJ, et al. (2017). “Neuroactive Steroids. 2. 3α-Hydroxy-3β-methyl-21-(4-cyano-1H-pyrazol-1′-yl)-19-nor-5β-pregnan-20-one (SAGE-217): A Clinical Next Generation Neuroactive Steroid Positive Allosteric Modulator of the (γ-Aminobutyric Acid)A Receptor”. Journal of Medicinal Chemistry. 60 (18): 7810–7819. doi:10.1021/acs.jmedchem.7b00846. PMID28753313.
^World Health Organization (2019). “International nonproprietary names for pharmaceutical substances (INN): recommended INN: list 82”. WHO Drug Information. 33 (3). hdl:10665/330879.
^Clayton AH, Lasser R, Parikh SV, Iosifescu DV, Jung J, Kotecha M, et al. (May 2023). “Zuranolone for the Treatment of Adults With Major Depressive Disorder: A Randomized, Placebo-Controlled Phase 3 Trial”. The American Journal of Psychiatry: appiajp20220459. doi:10.1176/appi.ajp.20220459. PMID37132201. S2CID258461851.
Clinical trial number NCT04442503 for “A Study to Evaluate the Efficacy and Safety of SAGE-217 in Participants With Severe Postpartum Depression (PPD)” at ClinicalTrials.gov
Clinical trial number NCT02978326 for “A Study to Evaluate SAGE-217 in Participants With Severe Postpartum Depression” at ClinicalTrials.gov
Vodobatinib (K0706) is a potent, third generation and orally active Bcr-Abl1 tyrosine kinase inhibitor with an IC50 of 7 nM. Vodobatinib exhibits activity against most BCR-ABL1 point mutants, and has no activity against BCR-ABL1T315I. Vodobatinib can be used for chronic myeloid leukemia (CML) research. Vodobatinib is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Vodobatinib (K0706) is a potent, third generation and orally active Bcr-Abl1 tyrosine kinase inhibitor with an IC50 of 7 nM. Vodobatinib exhibits activity against most BCR-ABL1 point mutants, and has no activity against BCR-ABL1T315I. Vodobatinib can be used for chronic myeloid leukemia (CML) research[1][2]. Vodobatinib is a click chemistry reagent, itcontains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
A mixture of methyl 3-iodo-4-methylbenzoate (2.0g, 7mmol), trimethylsilylacetylene (1.2ml, 8mmol), Pd(PPh3)4 (0.42g, 0.3mmol), CuI (0.137g, 0.7mmol) and diisopropylethylamine (2.5ml, 11.4mmol) in THF (20ml) was heated at 50°C for 12hrs under nitrogen atmosphere. The reaction mixture was cooled to ambient temperature and filtered through a Celite® bed. The clear filtrate was concentrated and the residue purified by flash chromatography on silica gel (elution with 2% ethyl acetate in n-hexane) to provide methyl 4-methyl-3-[(trimethylsilyl)ethynyl]benzoate.
To the solution of methyl 4-methyl-3-[(trimethylsilyl)ethynyl]benzoate (2.3g) in THF (40ml) was added tetrabutylammonium fluoride (1.0M in THF, 3.2ml, 1 1mmol) at ambient
temperature and stirred for 15 minutes, concentrated and the residue purified by flash chromatography on silica gel (elution with 2% ethyl acetate in n-hexane) to provide methyl 3 – ethynyl- 4-methylbenzo at e .
Similarly were prepared the following ester compounds from their corresponding iodo esters:
Methyl 3-ethynyl-4-fluorobenzoate
Methyl 3-ethynyl-4-methoxybenzoate
Reƒerence Example 2
4-Methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid
A mixture of methyl 3-ethynyl-4-methylbenzoate (0.341 g, 2mmol), 3-iodoquinoline (0.5g, 2mmol), Pd(PPh3)4 (0.1 1g, 0.01mmol), CuI (0.179g, 0.1mmol) and diisopropylethylamine (0.5ml, 3mmol) in DMF (15ml) was stirred at ambient temperature for 12hrs under an atmosphere of nitrogen. The reaction mixture was concentrated and the crude product was purified by flash chromatography on silica gel (elution with 10% ethyl acetate in n-hexane) to provide methyl 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoate.
Sodium hydroxide (0.15g, 3.71mmol) was added to a solution of the above methyl ester in methanol (20ml) and water (3ml) and stirred at 50°C for 3hrs and then concentrated in vacuo. Water (10ml) was added to the residue, adjusted pH to 4.0-4.5 with citric acid. The solid obtained was filtered, washed successively with water and diethyl ether and dried at ambient temperature to obtain 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid. 1H NMR (500 MHz in DMSO-d6), δ 2.66 (s, 3H), 7.56 (d, J = 8.0 Hz, 1H), 7.75 (t, J; = 15.1 Hz, J2 = 8.2 Hz, 1H), 7.89 (t, J} = 13.7 Hz, J2 = 8.5 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 8.2 Hz, 1H), 8.12 (d, J = 8.1 Hz, 1H), 8.17 (s, 1H), 8.75 (s, 1H), 9.1 1 (s, 1H), 12.84 (s, 1H).
Reƒerence Example 3
4-Methyl-3-[2-(3-quinolyl)ethynyl]benzohydrazide
A mixture of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid (0.15g, 0.5mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (0.15g, 0.7mmol) and 1-hydroxybenzotriazole (0.1g, 0.7mmol) in N,N-dimethylformamide (15ml) was stirred at room temperature for 1hr. Hydrazine hydrate (1.52ml, 0.5mmol) was then added and the mixture stirred for another 3hrs. Concentration and trituration of the residue with water produced a solid which was filtered, washed successively with water and diethyl ether, and finally dried in vacuo to get the hydrazide as a pale yellow solid.
N’-(3-iodo-4-methylbenzoyl)-2,4,6-trichlorobenzohydrazide was prepared by the reaction of 3-iodo-4-methylbenzoic acid with 2,4,6-trichlorobenzohydrazide. The coupling was performed in a manner similar to that described in Reference Example 3.
A mixture of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid (0.15g, 0.5mmol), N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (0.15g, 0.7mmol) and 1-hydroxybenzotriazole (0.1g, 0.7mmol) in N,N-dimethylformamide (15ml) was stirred at ambient temperature for 1hr. 2,4,6-Trichlorobenzohydrazide (0.125g, 0.5mmol) was added and the mixture stirred for 12hrs at ambient temperature. Concentration and trituration of the residue with water produced a solid which was filtered, washed with water and the crude product was purified by flash chromatography on silica gel (elution with 10% methanol in dichloromethane) to get 2,4,6-trichloro-N-[4-methyl-3-[2-(3-quinolyl)ethynyl]benzoyl] benzohydrazide as a white solid.
Method B:
2,4,6-Trichloro-N’-[4-methyl-3-[2-(3-quinolyl)ethynyl]benzoyl] benzohydrazide was also prepared by the reaction of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzoic acid with 2,4,6-trichlorobenzohydrazide in diethyl cyanophosphonate. The condensation reaction was performed in a manner similar to that described in Method A.
Method C:
2,4,6-Trichloro-N-[4-methyl-3-[2-(3-quinolyl)ethynyl]benzoyl]benzohydrazide was also prepared by the reaction of 4-methyl-3-[(quinolin-3-yl)ethynyl]benzohydrazide with 2,4,6- trichlorobenzoyl chloride. The condensation reaction was performed in a manner similar to that described in Method A.
The compounds 1.2 to 1.14, 1.21 to 1.34, 1.36 to 1.40, and 1.43 to 1.59 were prepared in a manner similar to Example I.1, by following either of the methods A, B or C, using the appropriate substrates.
Vodobatinib (N’-(2-chloro-6-methylbenzoyl)-4-methyl-3-[2-(3-quinolyl) ethynyl]-benzohydrazide), a c-Abl inhibitor is represented by Formula I (referred hereinafter interchangeably as vodobatinib or compound of Formula
International Publication Nos. WO 2017/208267A1, WO 2020/250133 Al and WO 2022/024072A1, which are hereby incorporated by reference, disclose methods of use of the compound of Formula I for the treatment of Parkinson’s disease, synucleinopathies and Alzheimer’s disease (AD) respectively.
There is a continuing need for effective and safe methods for the treatment of, and delaying the progression of, neurodegenerative diseases, including in the early-stage of the diseases.
Carebastine is the active metabolite of Ebastine. Carebastine is a histamine H1 receptor antagonist. Carebastine inhibits VEGF-induced HUVEC and HPAEC proliferation, migration and angiogenesis in a dose-dependent manner. Carebastine suppresses the expression of macrophage migration inhibitory factor.
Carebastine is the active metabolite of Ebastine. Carebastine is a histamine H1 receptor antagonist. Carebastine inhibits VEGF-induced HUVEC and HPAEC proliferation, migration and angiogenesis in a dose-dependent manner[1]. Carebastine suppresses the expression of macrophage migration inhibitory factor[2].
Literature References: Nonsedating type histamine H1-receptor antagonist. Prepn: J. M. P. Soto et al., EP 134124; eidem, US 4550116 (both 1985 to Fordonal). Metabolized in vivo to carebastine, its active carboxylic acid metabolite.
These schemes also illustrate the interrelatedness of the processes and intermediates.
EXAMPLE 1
One gram of 9 was dissolved in 20 mL of DMF and 18 mg of P(tBu)3, 41 mg of Pd(dba)2, 230 mg of ZnF2 and 1.2 g of 5 were added. A mixture was stirred at 80° for 18 hours, cooled to room temperature, diluted with ether and washed with water. The organic layer was dried over sodium sulfate, filtered and stripped in vacuo. The resulting product was flash chromatographed on silica gel using 4:1 hexane ethyl acetate to yield 1.0 g (91%) of 10. A repeat of the reaction on larger scale using 15 g of 9 provided 15.2 g (93%) of 10.
EXAMPLE 2
Five grams of 9 was dissolved in 50 mL of methylene chloride and cooled to 0° C. To the solution was added 5.78 g of trimethylsilyl iodide. The mixture was stirred for 30 minutes and excess sodium bisulfite solution was added with vigorous stirring at room temperature. The layers were separated and the aqueous layer extracted twice with methylene chloride. Combined organic layers were dried, filtered and stripped in vacuo to provide 7.7 g (98%) of 1. The reaction was repeated on a larger scale using 15 g of 9 to produce 22.5 g of 1 (96%) yield.
EXAMPLE 3
Six grams of potassium carbonate, 5.8 g of piperidine 2 and 7.6 g of 1 are combined in 100 mL of DMF. The suspension is stirred at room temperature until TLC in 4:1 hexane-ethyl acetate indicates a complete reaction. The reaction mixture is poured into 400 mL of water and extracted three times with methylene chloride. The combined organic extracts are dried, filtered and reduced in vacuo. The resulting product is flash chromatographed on silica gel using ethyl acetate containing 10% triethylamine to yield 3.
EXAMPLE 4
Seven grams of 3 is dissolved in 100 mL of methanol, cooled to 0° C. and 1.1 g of sodium borohydride is added. The mixture is stirred 1 hour, concentrated and partitioned between ethyl acetate and saturated aqueous sodium bicarbonate. The bicarbonate layer is extracted twice with ethyl acetate, the combined organic layers are dried over sodium sulfate and the solution is reduced in vacuo to provide 4.
EXAMPLE 5
Two grams of 4 is dissolved in 30 mL of DMF. To this are added 16.2 mg of P(tBu)3, 36.6 mg of Pd(dba)2, 209 mg of ZnF2 and 1.056 g of 5. The mixture is heated at 80° C., cooled, diluted with ether and worked up as in example 1. The resulting product is flash chromatographed on silica gel using 9:1 ethyl acetate-triethylamine to provide 7.
EXAMPLE 6
One hundred fifty milligrams of 6 is slurried in 5 mL of water and 10 mL of methanol. To the slurry is added 175 mg of sodium hydroxide. The slurry is refluxed for one hour, cooled to room temperature and the methanol removed in vacuo. The resulting aqueous solution is distributed between water and chloroform, the chloroform layer is discarded, the aqueous layer is adjusted to pH 2.3 and extracted with chloroform. The organic layer is dried, filtered and reduced in vacuo to provide carebastine.
EXAMPLE 7
Five grams of 1 was combined with 2.64 g of 2 and 2.0 g of potassium carbonate and 80 mL of DMF. The mixture was stirred at room temperature for two hours, poured into 400 mL of water and extracted three times into methylene chloride. The combined organic layers were dried, filtered and reduced in vacuo. The resulting product was flash chromatographed on silica gel using 9:1 ethyl acetate-triethylamine to provide 2.0 g (54%) of 3.
EXAMPLE 8
One and seven-tenths grams of 3, 90 mg of P(tBu)3, 300 mg of Pd(dba)2, 250 mg of ZnF2 and 1.1 g of 5 were dissolved in 330 mL of DMF under argon. The mixture was heated to 80° for two hours, cooled to room temperature, diluted with ether and worked up as described in example 1. The resulting product was filtered through silica to provide 1.2 g (67.8%) of 6.
EXAMPLE 9
Two grams of 20, 170 mg of P(tBu)3, 560 mg of Pd(acac)2, 474 mg of ZnF2 and 2.0 g of 5 were combined in 50 mL of DMF under argon. The mixture was heated to 80° C. and monitored by HPLC. When reaction was complete, the mixture was cooled to room temperature and 250 mL of water was added. The mixture was extracted three times with ether, dried, filtered and reduced in vacuo. The resulting product was flash chromatographed in 4:1 hexane-ethyl acetate to provide 1.89 g (85%) of 8.
EXAMPLE 10
Two grams of the triflate analog of 20 were reacted as in the foregoing example with 134 mg P(tBu)3, 433 mg of Pd(acac)2, 375 mg of ZnF2 and 1.58 g of 5 to provide 1.56 g (90% yield) of 8.
Example 11
Piperidinol 25 is reacted with chlorodiphenylmethane as described in Fujii et al. Arzneim.-Forsch. 44, 527-538 (1994) to provide 6.
Example 1: Potassium 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate (carristin potassium salt ) preparation
[0060]
[0061]
Step 1: Preparation of methyl 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate
[0062]
[0063]
Add 4-(diphenylmethoxy)piperidine hydrochloride (473mg, 1.77mmol), DMAC (4.5ml), K 3 PO 4 (1.13g, 5.3mmol), KI (29mg, 0.177mmol) to a 25ml single-neck bottle. , stir and heat to 100°C. Weigh 2-[4-(4-chloro-1-butyryl)phenyl]-2-methylpropionate methyl ester (600mg, 2.12mmol) and dissolve it in 1ml of DMAC. Add the reaction solution slowly and dropwise, and keep the reaction for 4~ 6h, TLC detects that the raw material reaction is complete. Cool to room temperature, add isopropyl acetate and water, and stir to separate layers. The aqueous phase was then extracted with isopropyl acetate, the organic phases were combined, washed twice with water, dried over anhydrous sodium sulfate, filtered, concentrated, and passed through a silica gel column to obtain 500 mg of the title product, yield 45%, purity: 97.3%.
Step 2: Preparation of 2-(4-(4-(4-(Diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (carristin)
[0067]
[0068]
Add (5-methyl-2-oxo-1,3-dioxo-4-yl)methyl-2-(4-(4-(4-(diphenylmethoxy))piperidine-1 to a 25ml three-necked flask) -Methyl)-butyryl)phenyl)-2-methylpropionate (320 mg, 0.62 mmol), 1.5 ml of methanol, 2 ml of 10% NaOH, heated to 60°C for 2 hours, and the TLC raw material reaction was completed. After the reaction is completed, cool to room temperature, concentrate to dryness, add EA, add hydrochloric acid to adjust the pH to 2~3, layer the layers, wash once with water, dry the organic phase, and concentrate to dryness to obtain 300 mg of the title product. Yield: 95%, purity 95.0%.
Add 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (499mg, 1mmol) and acetonitrile 3.5 to a 25ml three-necked flask. ml, heated to 60°C, added potassium hydroxide (56 mg, 1 mmol), stirred, cooled down, a white solid precipitated, filtered, and dried to obtain 500 mg of carristine potassium salt, with a yield of 90% and a purity of 98.67%.
Example 2: Sodium 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionate (carristine sodium salt ) preparation
[0077]
[0078]
In this example, the preparation method of 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid is the same as in Example 1.
[0079]
Add 2-(4-(4-(4-(diphenylmethoxy)piperidin-1-yl)butyryl)phenyl)-2-methylpropionic acid (499mg, 1mmol) and acetonitrile 3.5 to a 25ml three-necked flask. ml, heated to 60°C, added sodium hydroxide (40 mg, 1 mmol) and stirred for 1 hour, concentrated to dryness, added methyl tert-butyl ether and stirred, filtered, and dried to obtain 458 mg of carristin sodium salt, yield 85%, purity 96.98 %.
Tacaciclib is a CDK inhibitor, antineoplastic effect.
The present invention is directed to methods of preparation of compound of formula (I) that is useful for inhibiting Cyclin-dependent kinase 7 (CDK7) and for treating diseases or disorders mediated thereby.
CDK7, which complexes with cyclin H and RING-finger protein MAT1, phosphorylates the cell cycle CDKs in the activation of T-loop, to promote their activities (Fisher et al., Cell., Aug 26;78(4):713- 24, 1994). As such, it has been proposed that inhibiting CDK7 would provide a potent means of inhibiting cell cycle progression, which may be especially relevant given that there is compelling evidence from gene knockout studies in mice for lack of an absolute requirement for CDK2, CDK4 and CDK6 for the cell cycle at least in most cell types (M alumbres et al., Nature Cell Biology, 11, 1275 – 1276, 2009), whilst different tumors appear to require some, but they are independent of other interphase CDKs (CDK2, CDK4 , CDK6). Recent genetic and biochemical studies have confirmed the importance of CDK7 for cell cycle progression (Larochelle. et al., Mol Cell., Mar 23;25(6):839-50. 2007; Ganuza et al., EM BO J., May 30; 31(11): 2498-510, 2012).
Cyclin-dependent kinase 7 (CDK7) activates cell cycle CDKs and is a member of the general Transcription factor II Human (TFIIH). CDK7 also plays a role in transcription and possibly in DNA repair. The trimeric Cak complex CDK7/CyclinH/MATl is also a component of TFIIH, the general transcription/DNA repair factor IIH (Morgan, DO., Annu.Rev. Cell Dev. Biol. 13, 261-91, 1997). As a TFIIH subunit, CDK7 phosphorylates the CTD (Carboxy-Terminal-Domain) of the largest subunit of RNA polymerase II (pol II). The CTD of mammalian pol (II) consists of 52 heptad repeats with the consensus sequence 1 YSPTSPS 7 and the phosphorylation status of the Ser residues at positions 2 and 5 has been shown to be
important in the activation of RNAP-II indicating that it is likely to have a crucial role in the function of the CTD. CDK7, which primarily phosphorylates Ser-5 (PSS) of RNAP-II at the promoter as part of transcriptional initiation (Gomes et ah, Genes Dev. 2006 Mar 1; 20(5):601-12, 2006), in contrast with CDK9, which phosphorylates both Ser-2 and Ser-5 of the CTD heptad (Pinhero et al., Eur. J. Biochem., 271, pp. 1004-1014, 2004).
In addition to CDK7, other CDKs have been reported to phosphorylate and regulate RNA pol (II) CTD. The other CDKs include, Cdk9/ Cyclin T1 or T2 that constitute the active form of the positive transcription elongation factor (P-TEFb) (Peterlin and Price, Mol Cell., Aug 4; 23(3): 297-305,2006) and Cdkl2/Cyclin K and Cdkl3/Cyclin K as the latest members of RNAPII CTD kinases (Bartkowiak et al., Genes Dev., Oct 1 5;24(20):2303-16, 2010; Blazek et al., Genes Dev .Oct 15;25(20):2158-72, 2011).
Disruption of RNAP II CTD phosphorylation has been shown to preferentially effect proteins with short half-lives, including those of the anti-apoptotic BCL-2 family. (Konig et al., Blood, 1, 4307-4312, 1997; The transcriptional non-selective cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1; (Gojoet al., Clin. Cancer Res. 8, 3527-3538, 2002).
This suggests that the CDK7 enzyme complexes are involved in multiple functions in the cell: cell cycle control, transcription regulation and DNA repair. It is surprising to find one kinase involved in such diverse cellular processes, some of which are even mutually exclusive. It also is puzzling that multiple attempts to find cell cycle dependent changes in CDK7 kinase activity remained unsuccessful. This is unexpected since activity and phosphorylation state of its substrate, CDC2, fluctuate during the cell cycle. In fact, it is shown that cdk7 activity is required for the activation of both Cdc2/Cyclin A and Cdc2/Cyclin B complexes, and for cell division. (Larochelle, S. et al. Genes Dev 12,370-81, 1998). Indeed, flavopiridol, a non-selective pan-CDK inhibitor that targets CTD kinases, has demonstrated efficacy for the treatment of chronic lymphocytic leukemia (CLL), but suffers from a poor toxicity profile (Lin et al.,). 27, 6012-6018, 2009; Christian et al., Clin. Lymphoma Myeloma, 9, Suppl.
3, S179-S185, 2009).
International publication WO2016193939, which is incorporated herein by reference for all purposes describes CDK7 inhibitors and processes for the preparation thereof. Inhibitors of CDK7 are currently being developed for the treatment of cancer. For drug development, it is typically advantageous to employ individual stereoisomers as they exhibit marked differences in pharmacodynamic, pharmacokinetic, and toxicological properties.
Example- 1: Preparation of compound of formula (I)
Scheme-1: Preparation of KRM-A
Step-4
KRM-A 4
Step-1: Preparation of 2-(3-bromophenyl)-3-methylbutanoic acid (1)
2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3-bromophenyl) acetic acid (XA, 150 g, 0.69 mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mixture was stirred for 2h at -78 °C followed by a drop wise addition of isopropyl bromide (X B , 255 g, 2.07 mol) over a period of 30 min. The reaction mixture was stirred at room temperature overnight. Then, the reaction mixture was quenched with IN HC1 (pH 2) and the obtained product was extracted to ethyl acetate (500 mL x 3). The combined organic layer was washed with water followed by brine solution. The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford the crude compound which was purified by silica column by eluting with 0-10% ethyl acetate-hexane system to afford the title compound (150 g, 83% yield) , HPLC purity-96%. The compound of formula (1) can also be prepared by the procedure described in CN 110590747.
Step-2: Preparation of Compound 3
2-(3-bromophenyl)-3-methylbutanoic acid (1, 510 g, 1.98 mol) was dissolved in 30% of IP A in water (10.2 L; 3.06 L of IPA-7.14 L of water) and ( 1L\ 2i ?)-cyclohexane-1,2-diamine (2, 113 g, 0.9 mol) was added. The reaction mixture was stirred at room temperature for 10 min until the precipitation was observed, then was heated to 100 °C until the solution became clear and stirred at the same temperature for another 30 min. The reaction mixture was allowed to slowly reach room temperature for 8-12h. The obtained solid was filtered and washed with 500 mL of 30% IPA-water mixture and dried under vacuum to afford the compound 3 (620 g, wet).
Work up (for Chiral purity): Small portion (100 mg) of compound 3 was taken in DCM (2-3 mL) and was added IN HC1 (pH 2) at 0 °C until the clear solution was observed. The compound was extracted into DCM, dried over NaiSCL and the solvent was evaporated to afford the title compound as white solid (20 mg). Chiral HPLC was recorded for this sample and 20.6% of undesired isomer was observed in chiral HPLC.
In order to improve the chiral purity of the title compound, the recrystallization method was performed as described below.
Step-3: Recrystallization
The compound 3 (619.90 g) was taken in 30% of IP A in water (12.4 L), then the mixture was heated to 100 °C until the solution became clear and was stirred at the same temperature for another 30min. The reaction mixture was allowed to reach room temperature slowly for 8-12h.
The obtained solid was filtered and washed with 500mL 30% IPA-water and dried under vacuum to afford a desired compound (360g, wet).
Work up for analysis (for Chiral purity): Small portion (100 mg) from above compound was taken in DCM (2-3mL), was added IN HC1 (pH 2) at 0 °C until the clear solution was observed and the compound was extracted to DCM, dried over NaiSCL and the solvent was evaporated to afford title compound as white solid (35 mg). Chiral HPLC was recorded for this sample and 10.3% of undesired isomer was observed in chiral HPLC.
The recrystallization method was repeated for three more times by using 30% of IPA in water as per the aforesaid procedure to obtain the purity of greater than 98.50% ee along with 0.27% other isomer to afford 286 g of compound 4.
Step-4: Preparation of (S)-2-(3-bromophenyl)-3-methylbutanoic acid (KRM-A)
The compound 4 (286 g) was taken in DCM (1.3 L), then was added IN HC1 at 0 °C until the clear solution was observed, and the compound was extracted to DCM (500 mL x 2). The organic layer was separated, washed with brine solution (500 mL) and dried over NaiSCL. The solvent was evaporated from the reaction mixture to afford title compound as white solid (148 g, 60% yield). Chiral HPLC: 98.50%
Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-1H-pyrazol-3-yl)-3-methylbutanamide
Step-la: Preparation ofKRM-D
To a stirred solution of KRM-A (lOOg, O.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0°C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude (S)- 2-(3-bromophenyl)-3-methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step-lb: Preparation of compound of formula (II)
(5)-2-(3-bromophcnyl)-3-mcthylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of ieri-butyl 3-amino-5-cyclopropyl-1H-pyrazole- l-carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to reach RT and stirred overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HC1 (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL), brine solution (500 mL), dried over anhydrous NaiSCL , filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (S)-5-( 2-(3-bromophenyl)-3-methylbutanamido)-3-
cyclopropyl- lH-pyrazole- 1-carboxylate (compound of formula (IG)) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H) + , HPLC: 80.80%, retention time:15.89 min) . The crude product was taken as such for next step without further purification.
Step-1 c: Preparation of compound of formula (I)
To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-1H-pyrazole-1-carboxylate (180 g, 1,731 mol) in dioxane (360 mL ) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature. After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The obtained compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL), brine solution (300 mL), and dried over anhydrous NaiSCL . The organic layer was concentrated to obtain a crude (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula (G)) as a semi-solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The obtained solid was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (1 lOg, Yield: 78% over two steps). LCMS: m/z= 362 (M+H) + , HPLC: 97.66%, retention time: 24.10 min
Step-2: Preparation of (S, E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl) amino)-3-methyl-l-oxobutan-2- yl) phenyl) pyridin-2-yl)-4-morpholinobut-2-enamide (Compound of formula (I))
To a degassed solution of (5)-2-(3-bromophcnyl)-N-(5-cyclopropyl-1 H-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (E)-4 -morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)Ch.DCM (3.38 g, 0.0042 mol, and 0.03eq) was added to the reaction mixture and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite ® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and
extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulfate. The organic layer was concentrated under vacuum at 35-40°C to obtain crude title compound (~80g).
The crude compound of formula (I), (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20% aqueous NaiCCL solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulfate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), then the reaction mixture was stirred overnight at room temperature and filtered through Celite ® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, until absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (70g, HPLC purity: 92.70%, retention time: 15.65 min).
Work-up for improved chiral purity: The above compound of formula (I) was dissolved in ethylacetate (~30 vol, 2L) and washed with aqueous citric acid (2 times, 400 mL x 1 and 200mL x 1), aqueous NaHCCL solution (2%, 500 mL x 1) and aqueous NaCl solution (10%, 500 mL x 1). The combined organic layer was dried over sodium sulfate and filtered. The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound of formula (I) (~60g).
In some embodiments, the compound of formula (I) is (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide or a pharmaceutically acceptable salt or a stereoisomer thereof (Compound 44).
Compound 44 is disclosed in WO 2016/193939 Al, published December 8, 2016, entitled “Substituted heterocyclyl derivatives as cdk inhibitors,” the entire contents of which are incorporated herein by reference. Compound 44A can be in the form of a fumaric acid salt or cocrystal as described in WO 2022/130304 Al, published June 23, 2022, entitled “Cocrystal of a cdk inhibitor,” the entire contents of which are incorporated herein by reference.
Example 3: Synthesis of Compounds 44A & 44B via Chiral Separation
Scheme-1
Step-1: Synthesis of 2-(3-bromophenyl)-3-methylbutanoic acid
[0352] 2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3 -bromophenyl) acetic acid (reagent-1, 150g, 0.69mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mass was stirred for 2h at -78 °C followed by the drop wise addition of Isopropyl bromide (255 g, 2.07mol) over a period of 30 min at -78 °C. The reaction mass was stirred at room temperature for overnight. The reaction mass was quenched with IN HC1 (pH 2) and product extracted to ethyl acetate (500mL x 3). The combined organic layer washed with water followed by brine, dried and concentrated under reduced pressure to afford the title crude compound which was purified by silica column by eluting with 0-10% ethyl acetate -hexane system to afford the title compound 2 (150g, 83% yield). LCMS: m/z = 254.80 (M-2H)’
Step-2: Synthesis of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate
[0353] 2-(3-bromophenyl)-3-methylbutanoic acid (intermediate-2, 70g, 0.0.27mol) was dissolved in dry DCM (500 mL) and added oxalyl chloride (68 mL, 0.78mol) dropwise at 0 °C followed by addition of catalytic amount of DMF (0.8mL) and maintained reaction mass at same temperature for 30min. The reaction mass was allowed to room temperature and stirred for 4h, distilled off the solvent and excess oxalyl chloride under vacuum. Re-dissolved the residue in DCM (250 mL) and added slowly to the cooled solution of tert-butyl 3 -amino-5 -cyclopropyl- 1H-pyrazole-1 -carboxylate (intermediate-3, 49g, 0.218mol) and TEA (55 mL, 0.546mol) in THF (250 mL) at 0 °C for 30min, The reaction was stirred at room temperature for 12h then the reaction mass was concentrated under reduced pressure and the residue was dissolved in DCM, washed with saturated NaHCO3 solution and brine. The organic layer was dried over anhydrous sodium sulphate and concentrated under reduced pressure, the crude was purified by silica gel column chromatography by eluting with 15% ethyl acetate-hexane to afford the title compound 4 (90g, 71% ) LCMS: m/z = 363.80 (M-Boc+2).
Step-3: Synthesis of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl)butanamido)-l H -pyr azole- 1 -carboxylate
[0354] To a degassed solution of tert-butyl 3-(2-(3-bromophenyl)-3-methylbutanamido)-5-cyclopropyl-lH-pyrazole-1 -carboxylate (intermediate-4, 90g, 0.193mol) and 4, 4, 4′, 4′, 5, 5, 5′, 5′-octamethyl-2,2′-bi(l,3,2-dioxaborolane) (62g, 0.25 Imol) in 1,4-Dioxane (500 mL) was added potassium acetate (37.80g, 0.386mol). The reaction mass was allowed to stir for 10 min with degassing at RT and added PdC12(dppf).DCM complex (12.5g, 0.015mol). The reaction mass was heated for 3-4 h at 100 °C. Reaction mixture cooled to RT and filtered on celite bed, filtrate evaporated to get dark brown liquid. The crude material was purified by silica column chromatography by eluting with 20% ethyl acetate in hexane to afford the compound 5 (90g, 86%). LCMS: m/z = 410 (M-Boc+1)+.
Step-4: Synthesis of (E)-N-(5-(3-(l-((5-cyclopropyl-lH-pyrazol-3-yl)amino)-3-methyl-l-oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide
[0355] To a degassed solution of tert-butyl 5-cyclopropyl-3-(3-methyl-2-(3-(4, 4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)phenyl)butanamido)- 1 H-pyrazole- 1 -carboxylate, 5 (10g, 0.019mol) and (E)-N-(5-bromopyridin-2-yl)-4-morpholinobut-2-enamide (7.7g, 0.023mol) in
1,4-Dioxane (lOOmL) and water (40mL) followed by Cs2CO3 (14.5g, 0.045mol) were added. The reaction mass was allowed to stir for 10 min with degassing and added Pd(PPh3)4 (1.1g, 0.00095mol), heated the reaction mass for 4 h at 100 °C in a sealed tube. The reaction mass was cooled and diluted with brine solution. The aqueous layer was separated and re-extracted with ethyl acetate. The combined organic layer was evaporated to dryness and crude material was purified by silica column chromatography by eluting with 10%-l 5 % methanol in DCM to get desired pure compound 44 (4.5g, 44%). LCMS: m/z = 529.15 (M+H)+; HPLC: 95.17%, rt: 6.34 min.
[0356] Racemic (E)-N-(5 -(3 -( 1 -((5 -cyclopropyl- 1 H-pyrazol-3 -yl)amino)-3 -methyl- 1 -oxobutan-2-yl)phenyl)pyridin-2-yl)-4-morpholinobut-2-enamide was separated by using chiral preparative HPLC column (Method: Column: Chiral Pak IA (20mm X 250 mm, 5 micron), Elution: isocratic (50:50), A=ACN, B= MeOH, Flow: 20mL/min ) to afford the pure Isomer- 1 and Isomer-2.
Example 4: Preparation of Compound 44-A via Chiral Synthesis
Preparation of KRM-A (chemical precursor to Compound 44-A)
Step-4
KRM-A
Step-1: Preparation of 2-(3-bromophenyl)-3-methylbutanoic acid (1)
[0359] 2M LDA (698 mL, 1.38mol) was added to a solution of 2-(3 -bromophenyl) acetic acid (150 g, 0.69 mol) in THF (700mL) at -78 °C over a period of 30 min. The reaction mixture was stirred for 2h at -78 °C followed by drop wise addition of isopropyl bromide (XB, 255 g, 2.07 mol) over a period of 30 min at -78 °C. The reaction mass was stirred at room temperature overnight. The reaction mass was quenched with IN HC1 (pH 2) and the obtained product was extracted to ethyl acetate (500 mL x 3). The combined organic layer was washed with water followed by brine, dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford the title crude compound which was purified by silica column by eluting with 0-10% ethyl acetate -hexane system to afford the title compound (150 g, 83% yield), HPLC purity-96%. The compound of formula (1) can also be prepared by the procedure described in CN110590747.
Step-2: Preparation of Compound 3
[0360] 2-(3-bromophenyl)-3-methylbutanoic acid (1, 510 g, 1.98 mol) was dissolved in 30% of IPA in water (10.2 L; 3.06 L of IPA-7.14 L of water) and (1R, 27?)-cyclohexane-l,2-diamine (2, 113 g, 0.9 mol) was added. The reaction mixture was stirred at room temperature for 10 min until the precipitation was observed, then heated to 100 °C till the solution becomes clear and was stirred at same temperature for another 30 min. The reaction mixture was allowed to attain room temperature slowly for 8-12h. The obtained solid was filtered and washed with 500 mL of 30% IPA-water mixture and dried under vacuum to afford the compound 3 (620 g, wet).
[0361] Work up for analysis (for Chiral purity): Small portion (100 mg) of compound 3 was taken in DCM (2-3 mL) and was added IN HC1 (pH 2) at 0 °C till the clear solution was observed. The compound was extracted into DCM, dried over Na2SC>4 and the solvent was evaporated to afford the title compound as white solid (20 mg). Chiral HPLC was recorded for this sample and 20.6% of undesired isomer was observed in chiral HPLC.
[0362] In order to improve the chiral purity of the title compound, the recrystallization method was performed as described below.
Step-3: Recrystallization
[0363] The compound 3 (619.90 g) was taken in 30% of IPA in water (12.4 L), then the mixture was heated to 100 °C till the solution becomes clear and stirred at same temperature for another 30min. The reaction mixture was allowed to attain room temperature slowly for 8-12h. The obtained solid was filtered and washed with 500mL 30% IP A- water and dried under vacuum to afford a desired compound (360g, wet).
[0364] Work up for analysis (for Chiral purity): Small portion (100 mg) from above compound was taken in DCM (2-3mL), was added IN HC1 (pH 2) at 0 °C till the clear solution was observed and the compound was extracted to DCM, dried over Na2SCL and the solvent was evaporated to afford title compound as white solid (35 mg). Chiral HPLC was recorded for this sample and 10.3% of undesired isomer was observed in chiral HPLC.
[0365] The recrystallization method was repeated for three more times by using 30% of IPA in water as described above to get the purity >98.50% ee along with 0.27% other isomer to afford 286 g of compound 4.
Step-4: Preparation of (S)-2-(3-bromophenyl)-3-methylbutanoic acid (KRM-A)
[0366] The compound 4 (286 g) was taken in DCM (1.3 L), then was added IN HC1 at 0 °C until the clear solution was observed, and the compound was extracted to DCM (500 mL x 2). The organic layer was separated and washed brine solution (500 mL) and dried over Na2SO4, the solvent was evaporated to afford title compound as white solid (148 g, 60% yield). Chiral HPLC: 98.50%
Step-1: Synthesis of (S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylhutanamide
Step- la: Preparation of KRM-D
[0368] To a stirred solution of KRM-A (100g, 0.388mol) in dry DCM (600 mL, 6 vol), a catalytic amount of DMF (10 mL) was added followed by oxalyl chloride (45 mL, 0.525 mol) dropwise at 0 °C over a period of 30 min. After completion of addition, the reaction mixture was stirred for 15 min at the same temperature. The reaction mixture was allowed to reach room temperature and stirred for 2 to 4h. After completion of the reaction (reaction was monitored by TLC, acid chloride formation was checked by quenching an aliquot of reaction mixture with MeOH), the reaction mixture was concentrated under vacuum at 40°C-45°C to afford crude 2-(3-bromophenyl)-3 -methylbutanoyl chloride (KRM-D). The crude KRM-D was dissolved in toluene (500mL) and used for next step.
Step- lb: Preparation of compound of formula Z
[0369] (S)-2-(3-bromophenyl)-3 -methylbutanoyl chloride in toluene was added slowly to a pre-cooled solution (0 to 5 °C) of te/7-butyl 3 -amino-5 -cyclopropyl- IH-pyrazole-l -carboxylate (KRM-B, 95.5g, 0.427 mol) and N, N-diisopropylethyl amine (100 mL, 0.583 mol) in toluene (1.2 L) at 0 °C for the period of l-2h. The reaction mixture was allowed to attain RT and stirred for overnight. The reaction mixture was then cooled to 0-5°C and washed with ice-cold 1.5N HCI (3 x 500 mL). The organic layer was washed with sodium bicarbonate solution (500 mL),
brine solution (500 mL), dried over anhydrous Na2SO4, filtered and concentrated under vacuum at 45-50°C to afford crude tert-butyl (5)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (compound of formula Z) as light brown oil (~180g, LCMS: m/z= 461.9 (M+H)+, HPLC: 80.80%, retention time: 15.89 min). The crude product was taken as such for next step without further purification.
Step-lc: Preparation of compound of formula Y
[0370] To a suspension of tert-butyl (S)-5-(2-(3-bromophenyl)-3-methylbutanamido)-3-cyclopropyl-lH-pyrazole-1 -carboxylate (180 g, 1.731 mol) in dioxane (360 mL) was added 2N aqueous HC1 (360 mL) at 0 °C. The reaction mixture was stirred overnight at room temperature.
[0371] After completion of the reaction, dioxane was concentrated, and the reaction mixture was diluted with water (500 mL) and basified with solid sodium bicarbonate (until pH-8). The resulted compound was extracted with DCM (700 mL x 3). The combined organic layers were washed with water (300 mL) and brine solution (300 mL), and dried over anhydrous Na2SO4. The organic layer was concentrated to get a crude (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (Compound of formula Y) as a semi solid. The crude was dissolved in toluene (500 mL) and the solution was stirred for 18 h. The solid formed was filtered and washed with toluene (100 mL) and n-heptane (200 mL). The solid was further dried under vacuum at 45-50°C for 6 h to afford a title compound (110g, Yield: 78% over two steps). LCMS: m/z= 362 (M+H)+, HPLC: 97.66%, retention time: 24.10 min
[0373] To a degassed solution of (<S)-2-(3-bromophenyl)-N-(5-cyclopropyl-lH-pyrazol-3-yl)-3-methylbutanamide (50 g, 0.138 mol) and (£)-4-morpholino-N-(5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridin-2-yl)but-2-enamide (KRM-C, 56.6 g, 0.151 mol, 1.1 eq) (prepared according to the procedure described in W02020202001) in 1,4-dioxane (500 mL, 10 vol) and water (100 mL, 2 vol) was added K3PO4 tribasic (73.2 g, 0.345 mol, 2.5 eq) at room temperature The reaction mass was stirred for 20 min with argon purging (degassing). Pd(dppf)C12.DCM [l,l’-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with dichloromethane] (3.38 g, 0.0042 mol, and 0.03eq) was added and the reaction mixture was heated to 90°C for 1-2 h (The reaction was monitored by TLC using 10% methanol in DCM as solvent system).
[0374] After completion of the reaction, the reaction mass was cooled to room temperature and filtered through Celite® bed. The bed was washed with 1, 4-dioxane (200 mL) and the filtrate was concentrated to get crude compound. The crude compound was dissolved in 5% methanol in DCM (400 mL) and washed with water (200 mL x 2). The aqueous layer was separated and extracted with DCM (100 mL x 2). The combined organic layer was washed with brine solution, filtered and dried over sodium sulphate. The organic layer was concentrated under vacuum at 35-40°C to get crude title compound (~80g).
[0375] The crude compound 44A, (80 g) was dissolved in 700 mL of ethyl acetate. The reaction mixture was cooled to 15°C and 2N HC1 was slowly added (until pH ~1). The reaction mixture was then stirred at room temperature for 20 min and the layers were separated. The aqueous layer (containing the product) was washed with ethyl acetate (300 mL x 3). The aqueous layer was cooled to 0°C and adjusted the pH to ~8 using 20 % aqueous Na2COs solution. The product was extracted with 10% methanol in DCM (300 mL x 3). The combined organic layer was washed with water (300 mL), dried over sodium sulphate and filtered. The filtrate was treated with activated charcoal (16 g, 20% w/w with respect to crude input of 80 g), stirred overnight at room temperature and filtered through Celite® bed. The bed was washed with 5% methanol in DCM (~ 20 vol, till absence of product by TLC). The filtrate was concentrated under vacuum at 35°C – 40°C to afford compound 44A (70g, HPLC purity: 92.70%, retention time: 15.65 min).
Sotuletinib, also known as BLZ945, is a potent and selective CSF-1R kinase inhibitor. BLZ945 showed effects of CSF1R inhibition on other tumor-infiltrating immune cells. BLZ945 attenuates the turnover rate of TAMs while increasing the number of CD8+ T cells that infiltrate cervical and breast carcinomas. BLZ945 decreases the growth of malignant cells in the mouse mammary tumor virus-driven polyomavirus middle T antigen (MMTV-PyMT) model of mammary carcinogenesis. BLZ945 prevents tumor progression in the keratin 14-expressing human papillomavirus type 16 (K14-HPV-16) transgenic model of cervical carcinogenesis.
Sotuletinib (BLZ945) is an experimental drug in development for the treatment of amyotrophic lateral sclerosis (ALS). It works as a colony-stimulating factor 1 (CSF1) receptor inhibitor.[1][2][3]
OriginatorCelgene Corporation; Novartis
ClassAmides; Amines; Antineoplastics; Benzothiazoles; Cyclohexanols; Ethers; Pyridines; Small molecules
Mechanism of ActionMacrophage colony stimulating factor receptor antagonists
Phase IIAmyotrophic lateral sclerosis
Phase I/IISolid tumours
05 Dec 2022Novartis Pharmaceuticals terminates a phase I/II trials in Solid tumours (Combination therapy, Late-stage disease, Metastatic disease) in Taiwan, Japan, Israel (PO) in US, Israel, Italy, Japan, Singapore, Spain, Taiwan and Switzerland (EudraCT2015-005806-12) (NCT02829723)
14 Feb 2022Adverse events and pharmacodynamics data from preclinical macaque model study in brain disorders presented at the 29th Conference on Retroviruses and Opportunistic Infections
03 Dec 2020Chemical structure information added
An orally bioavailable inhibitor of colony stimulating factor 1 receptor (CSF-1R; CSF1R), with potential antineoplastic activity. CSF1R inhibitor BLZ945 selectively binds to CSF1R expressed on tumor-associated macrophages (TAMs), blocks the activity of CSF1R, and inhibits CSF1R-mediated signal transduction pathways. This inhibits the activity and proliferation of TAMs, and reprograms the immunosuppressive nature of existing TAMs. Altogether, this reduces TAM-mediated immune suppression in the tumor microenvironment, re-activates the immune system, and improves anti-tumor cell responses mediated by T-cells. CSF1R, also known as macrophage colony-stimulating factor receptor (M-CSFR) and CD115 (cluster of differentiation 115), is a cell-surface receptor for its ligand, colony stimulating factor 1 (CSF1); this receptor is overexpressed by TAMs in the tumor microenvironment, and plays a major role in both immune suppression and the induction of tumor cell proliferation.
PATENT
The free base and salts of the compound of formula (I) may be prepared for example, according to the procedures given in International Patent Application No. PCT/US2007/066898 filed on Apr. 18, 2007 and published as WO2007/121484 on Oct. 25, 2007. The compound of formula (I) has the chemical name: 4-(2-((1R,2R)-2-hydroxycyclohexylamino)benzothiazol-6-yloxy)-N-methylpicolinamide and is also known as BLZ945.
Step 1. Preparation of 4-(2-((lR,2R)-2-aminocyclohexylamino)benzo[d]thiazol-6-yloxy)-N-methylpicolinamide To the solution of N-methyl-4-(2-(methylsulfinyl)benzo[d]thiazol-6-yloxy)picolinamide (15 mg, 43 μmole) in 400 μL of NMP was added (lR,2R)-cyclohexane-1,2-diamine (17 mg, 150 μmole). The reaction solution was stirred at 105°c for 24 hours. The crude reaction solution was purified on prep HPLC and evaporated in vaccuo to give 4-(2-((lR,2R)-2-aminocyclohexylamino)benzo[d]thiazol-6-yloxy)-N-methylpicolinamide (12 mg, 30 μmole) as white powder. ES/MS m/z 398.1(MH+).
^ Martinez-Gonzalez, Loreto; Martinez, Ana (1 February 2023). “Emerging clinical investigational drugs for the treatment of amyotrophic lateral sclerosis”. Expert Opinion on Investigational Drugs. 32 (2): 141–160. doi:10.1080/13543784.2023.2178416.
useful in the treatment of a steroid receptor, in particular androgen receptor (AR), dependent conditions and diseases, and to pharmaceutical compositions containing such compounds.
Prostate cancer is worldwide the most common cancer in men. Even though the 5-year survival rate of patients with localized prostate cancer is high, the prognosis for those patients, who develop castration-resistant prostate cancer (CRPC) within that 5-year follow-up period, is poor.
The androgen receptor (AR) signalling axis is critical in all stages of prostate cancer. In the CPRC stage, disease is characterized by high AR expression, AR amplification and persistent activation of the AR signalling axis by residual tissue/tumor androgens and by other steroid hormones and intermediates of steroid biosynthesis. Thus, treatment of advanced prostate cancer involves androgen deprivation therapy (ADT) such as hormonal manipulation using gonadotropin-releasing hormone (GnRH) agonists/antagonists or surgical castration, AR antagonists or CYP17A1 inhibitors (such as abiraterone acetate in combination with prednisone).
Although therapies can initially lead to disease regression, eventually majority of the patients develop a disease that is refractory to currently available therapies. Increased progesterone levels in patients treated with abiraterone acetate has been hypothesized to be one of the resistance mechanisms. Several nonclinical and clinical studies have indicated upregulation of enzymes that catalyse steroid biosynthesis at the late stage of CRPC. Very recently it has been published that 11β-OH androstenedione can be
metabolized into 11-ketotestosterone (11-K-T) and 11-ketodehydrotestosterone (11-K-DHT) which can bind and activate AR as efficiently as testosterone and dihydrotestosterone. It has been shown that these steroids are found in high levels in plasma and tissue in prostate cancer patients, suggesting their role as AR agonists in CRPC. Furthermore, it has been addressed that prostate cancer resistance to CYP17A1 inhibition may still remain steroid dependent and responsive to therapies that can further suppress de novo intratumoral steroid synthesis upstream of CYP17A1, such as by CYP11A1 inhibition therapy (Cai, C. et al, Cancer Res., 71(20), 6503-6513, 2011).
Cytochrome P450 monooxygenase 11A1 (CYP11A1), also called cholesterol side chain cleavage enzyme, is a mitochondrial monooxygenase which catalyses the conversion of cholesterol to pregnenolone, the precursor of all steroid hormones. By inhibiting CYP11A1, the key enzyme of steroid biosynthesis upstream of CYP17A1, the total block of the whole steroid biosynthesis can be achieved. CYP11A1 inhibitors may therefore have a great potential for treating steroid hormone dependent cancers, such as prostate cancer, even in advanced stages of the disease, and especially in those patients who appear to be hormone refractory. It has been recently shown that a compound having CYP11A1 inhibitory effect significantly inhibited tumor growth in vivo in a murine CRPC xenograft model (Oksala, R. et al, Annals of Oncology, (2017) 28 (suppl.
To a solution of 5-hydroxy-2-(isoindolin-2-ylmethyl)-4H-pyran-4-one (0.10 g, 0.41 mmol) in DMF (2 ml) were added (4-(methylsulfonamidomethyl)cyclohexyl)methyl methanesulfonate (0.14 g, 0.45 mmol) and K2CO3 (0.12 g, 0.8 mmol). The reaction mixture was heated at 80 °C for 2 h. The mixture was cooled to RT, water (10 ml) was added and the product was extracted with EtOAc. The combined extracts were washed with water, dried with Na2SO4, filtered and evaporated. The crude product was purified by column chromatography to afford the title compound (0.06 g). 1H NMR (400 MHz, Chloroform-d) δ ppm 0.92 – 1.11 (m, 4 H) 1.40 – 1.63 (m, 2 H) 1.78 – 2.00 (m, 4 H) 2.91 – 2.99 (m, 5 H) 3.65 (d, J=6.46 Hz, 2 H) 3.77 (s, 2 H) 4.03 (s, 4 H) 5.04 (br t, J=6.31 Hz, 1 H) 6.49 (s, 1 H) 7.20 (s, 4 H) 7.59 (s, 1 H).
To a stirred solution of 2-(chloromethyl)-5-hydroxy-4H-pyran-4-one (2.0 g, 12.5 mmol) in acetonitrile (50 mL) were added DIPEA (3.22 mL, 25.0 mmol) and isoindoline (1.78 g, 25.0 mmol) at RT. When the reaction was complete, the precipitated solid was filtered and washed with EtOAc. The title compound was collected as pale brown solid (1.1 g). LC-MS: m/z 244.1 (M+H)+.
///////////////////
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
The rising global epidemic of obesity and its comorbidities, e.g., type 2 diabetes mellitus and hyperlipidemia, is placing an enormous burden both on public health (mortality and morbidity) and on the available public health resources required to treat these conditions.
Current drugs that treat hyperlipidemia (e.g., statins, omega-3 fatty acids, fibrates) have mostly neutral effects on glycemic control, whilst drugs targeting glycemic control e.g., insulin, thiazolidinediones (TZDs), have adverse effects upon bodyweight and (for TZDs) other unwanted side-effects restricting their use.
In addition to hyperlipidemia and type 2 diabetes, a marked increase in the prevalence of non-alcoholic fatty liver disease (NAFLD) has occurred. NAFLD has become the most common chronic liver condition in Western populations in relation to the obesity and type 2 diabetes epidemics. The prevalence of non-alcoholic steatohepatitis (NASH), a form of NAFLD that is associated with hepatic inflammation and ballooning of hepatocytes, is expected to increase by 63% between 2015 and 2030 in the United States (Estes, Hepatology, 2018; 67(1): 123-133), where NASH is expected to become the leading cause of liver transplantation by 2020. As liver fibrosis, but not inflammation, is associated with mortality and morbidity in NASH patients, drugs which prevent progression/induce regression of fibrosis are also a focus of biomedical research.
The development of novel compounds that simultaneously target both hyperlipidemia and glycemic control, without the adverse side-effects (e.g., weight gain) typically associated with insulin sensitising drugs is thus a desirable goal. Such compounds would be even more attractive if they could additionally prevent the progression/reverse hepatic fibrosis and reduce hepatic steatosis. The present invention addresses these needs for new treatment methods, compounds, and pharmaceutical compositions.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
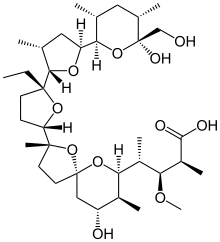
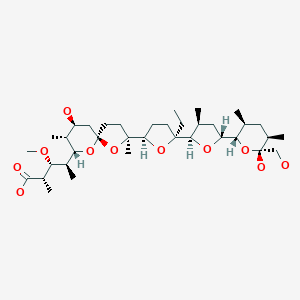









 verify (what is ?)Infobox references
verify (what is ?)Infobox references

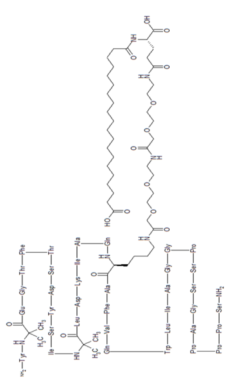







 (tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes
(tirzepatide) injection, the first and only GIP and GLP-1 receptor agonist for the treatment of adults with type 2 diabetes