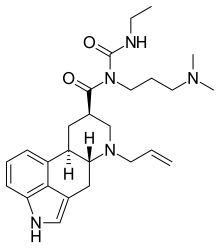
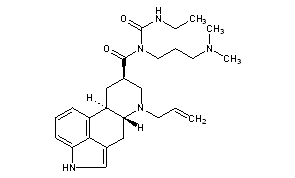
Cabergoline
Cabaser
- Molecular FormulaC26H37N5O2
- Average mass451.604 Da
1-Ethyl-3-(3′-dimethylaminopropyl)-3-(6′-allylergoline-8’β-carbonyl)urea
5860
81409-90-7[RN]
(6aR,9R,10aR)-N-[3-(dimethylamino)propyl]-N-(ethylcarbamoyl)-7-prop-2-en-1-yl-4,6,6a,7,8,9,10,10a-octahydroindolo[4,3-fg]quinoline-9-carboxamide
(8b)-N-[3-(Dimethylamino)propyl]-N-[(ethylamino)carbonyl]-6-(2-propenyl)ergoline-8-carboxamide
(8β)-6-Allyl-N-[3-(dimethylamino)propyl]-N-(ethylcarbamoyl)ergoline-8-carboxamide
Ergoline-8-carboxamide, N-[3-(dimethylamino)propyl]-N-[(ethylamino)carbonyl]-6-(2-propen-1-yl)-, (8β)-
ergoline-8-carboxamide, N-[3-(dimethylamino)propyl]-N-[(ethylamino)carbonyl]-6-(2-propenyl)-, (8β)-
KE6167600, LL60K9J05T, UNII-LL60K9J05T,
каберголин
كابارغولين
卡麦角林,
- FCE 21336
- FCE-21336
Cabergoline
CAS Registry Number: 81409-90-7
CAS Name:(8b)-N-[3-(Dimethylamino)propyl]-N-[(ethylamino)carbonyl]-6-(2-propenyl)ergoline-8-carboxamide
Additional Names: 1-ethyl-3-(3¢-dimethylaminopropyl)-3-(6¢-allylergoline-8¢b-carbonyl)urea; 1-[(6-allylergoline-8b-yl)carbonyl]-1-[3-(dimethylamino)propyl]-3-ethylurea
Manufacturers’ Codes: FCE-21336
Trademarks: Dostinex (Pharmacia & Upjohn)
Molecular Formula: C26H37N5O2, Molecular Weight: 451.60
Percent Composition: C 69.15%, H 8.26%, N 15.51%, O 7.09%
Literature References: Dopamine D2-receptor agonist. Prepn: P. Salvati et al.,BE888243; eidem,US4526892 (1981, 1985 both to Farmitalia Carlo Erba). Prepn and bioactivity: E. Brambilla et al.,Eur. J. Med. Chem.24, 421 (1989). Clinical pharmacology: C. Ferrari et al.,J. Clin. Endocrinol. Metab.63, 941 (1986). Veterinary trial as abortifacient in dogs: K. Post et al.,Theriogenology29, 1233 (1988). Clinical evaluation to prevent puerperal lactation: G. B. Melis et al.,Obstet. Gynecol.71, 311 (1988); in hyperprolactinemic disorders: C. Ferrari et al.,J. Clin. Endocrinol. Metab.68, 1201 (1989). Clinical trial in Parkinson’s disease: J. T. Hutton et al.,Neurology46, 1062 (1996).
Properties: White crystals from diethyl ether, mp 102-104°. Sol in ethyl alcohol, chloroform, DMF; slightly sol in 0.1 N HCl; very slightly sol in n-hexane. Insol in water. LD50 orally in male mice: >400 mg/kg (Brambilla).
Melting point: mp 102-104°
Toxicity data: Sol in ethyl alcohol, chloroform, DMF; slightly sol in 0.1 N HCl; very slightly sol in n-hexane. Insol in water. LD50 orally in male mice: >400 mg/kg (Brambilla)
Cabergoline diphosphate
85329-89-1
Derivative Type: Diphosphate
CAS Registry Number: 85329-89-1, Molecular Formula: C26H37N5O2.2H3PO4, Molecular Weight: 647.59
Percent Composition: C 48.22%, H 6.69%, N 10.81%, O 24.71%, P 9.57%
Properties: mp 153-155°., Melting point: mp 153-155°
Therap-Cat: Prolactin inhibitor; antiparkinsonian.
Therap-Cat-Vet: Prolactin inhibitor.
Keywords: Antiparkinsonian; Dopamine Receptor Agonist; Prolactin Inhibitor.
Cabergoline, sold under the brand name Dostinex among others, is a dopaminergic medication used in the treatment of high prolactin levels, prolactinomas, Parkinson’s disease, and for other indications. It is taken by mouth.
Cabergoline is an ergot derivative and a potent dopamine D2 receptor agonist.[1]
Cabergoline was patented in 1980 and approved for medical use in 1993.[2]
Cabergoline is a dopamine receptor agonist used for the treatment of hyperprolactinemic conditions due to various causes.
Cabergoline, an ergot derivative, is a long-acting dopamine agonist and prolactin inhibitor. It is used to treat hyperprolactinemic disorders and Parkinsonian Syndrome. Cabergoline possesses potent agonist activity on dopamine D2 receptors.
Synthesis Reference
PAPER
https://www.researchgate.net/publication/11103403_A_Practical_Synthesis_of_Cabergoline
N-[[(5R,8R,10R)-6-Allylergolin-8-yl]carbonyl]-N-[3-(di-methylamino)propyl]-N′-ethylurea (1). Compound 11 (40.13g, 72.7 mmol), H2O (200 mL), and 1 M aqueous hydrochloric acid(182 mL, 182 mmol) were combined and heated to 80 °C for 1 h.EtOAc (300 mL) was added to the light yellow solution. The pHwas adjusted to 10 with concentrated NH4OH (30 mL). Theorganic phase was separated and extracted with H2O(2×100mL). The aqueous layer was extracted with EtOAc (1 ×100 mL),and the combined organic layer was dried over Na2SO4, filtered,and concentrated to an amorphous solid. Cabergoline wasisolated in 94% yield (55% chemical yield overall from 8in highpurity (99 area % by HPLC): 1H NMR (CDCl3)δ1.18 (t, J)7.1 Hz, 3H), 1.76 (m, 2H), 1.85 (m, 2H), 2.23 (s, 6H), 2.34 (m,2H), 2.53-2.84 (m, 4H), 2.98 (m, 1H), 3.17 (m, 1H), 3.29-3.44(m, 4H), 3.55 (m, 1H), 3.83 (m, 2H), 5.19 (d, J)10.2 Hz, 1H),5.25 (d, J)16.8 Hz, 1H), 5.95 (m, 1H), 6.87 (m, 2H), 7.14 (m,2H), 8.88 (s, 1H), 9.45 (s, 1H); 13C NMR (100 MHz, CDCl3)δ14.7, 26.61, 31.31, 35.41, 40.04, 42.23, 43.21, 44.93, 55.61, 56.10,63.74, 108.7, 111.58, 113.10, 117.88, 118.36, 122.98, 126.02,132.67, 133.31, 133.83, 177.89.
1H AND 13 NMR IN CDCl3file:///C:/Users/Inspiron/Downloads/jo0203847_si_001.pdf
in dmso d6
SYN
DOI: 10.1016/0223-5234(89)90087-1

PATENT
https://patents.google.com/patent/EP1720869B1/en
- 6-Allyl-N-[3-(dimethylamino)propyl]-N-[(ethylamino)carbonyl]-ergoline-8β-carboxamide – international non-proprietary name cabergoline – of formula (I)is a potent dopamine agonist and is useful as anti-Parkinson drug and as prolactin inhibitor (Eur. J. Med. Chem. 1989, 24, 421-426 and United States Patent 5,382,669 ).
- [0003]
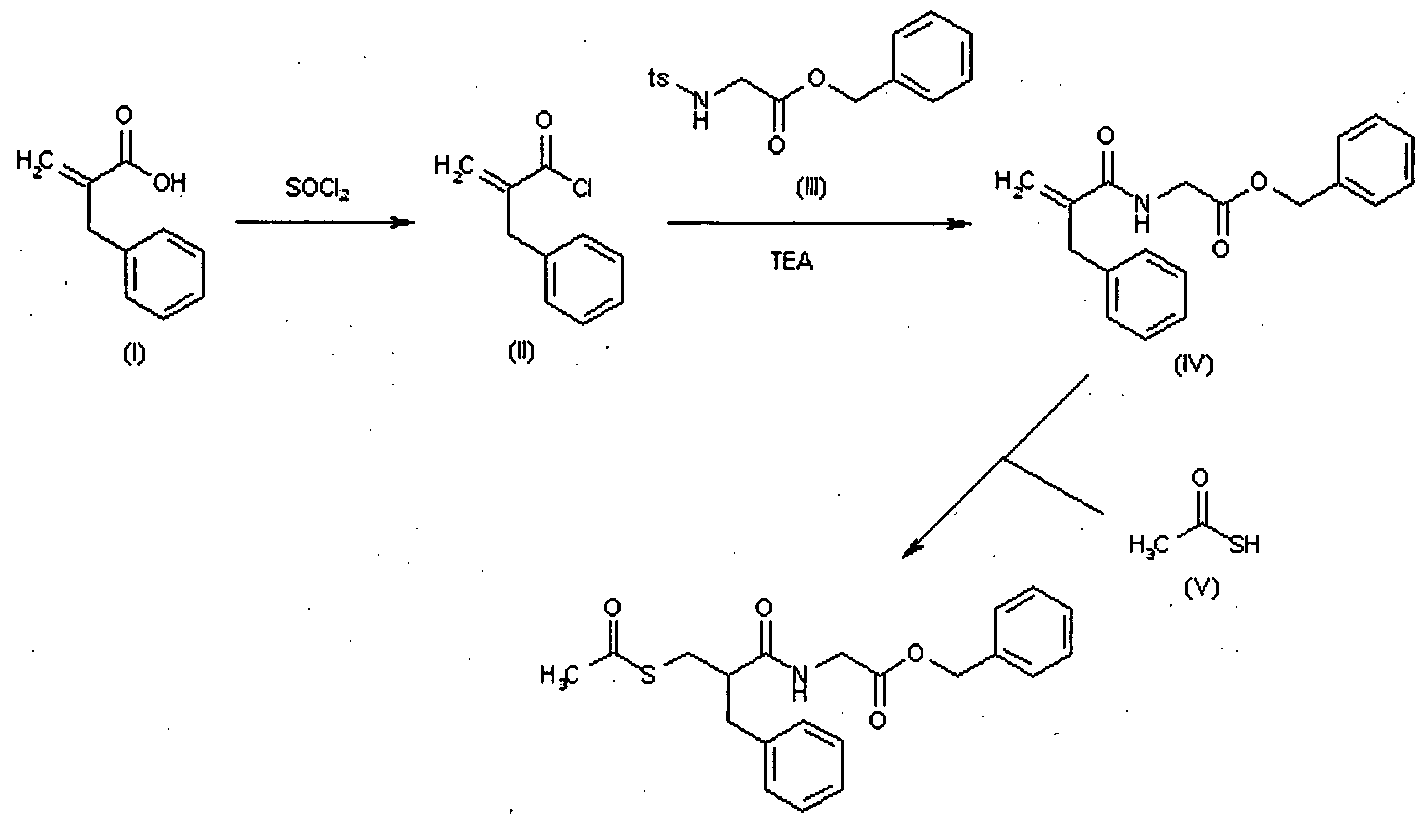
Cabergoline (I) was firstly prepared according to United States Patent 4,526,892 by reaction of 6-allyl-ergoline-8β-carboxylic acid (II) with 1-[3-(dimethylamino)propyl)-3-ethylcarbodiimide (EDC) (Scheme 1). - [0004]
In this case both regioisomers (I) and (III) were obtained and the yield of the isolated cabergoline (I) is only approx. 21% as a consequence of isolation difficulties, considering that the yield of compound (II) prepared from (XIII) according to the state of the art is 70%. - [0005]
Eur. J. Med. Chem. 1989, 24, 421-426 describes another method for the preparation of Cabergoline (I), which is based on the direct reaction of 6-allyl-N-[3-(dimethylamino)propyl]-ergoline-8β-carboxamide (IV) with ethyl isocyanate (EtNCO) (Scheme 2). - [0006]
Since this reaction leads to equilibrium, it requires the use of a large excess of ethyl isocyanate (up to 40 equivalents) for reasonable conversion and must be conducted at above 100°C in toluene for several hours. The use of large quantities of toxic ethyl isocyanate under drastic reaction conditions presents a serious hazard for the large-scale preparation of cabergoline (I). In addition, conversion to (I) is incomplete and competitive acylation of the indole nitrogen forming compounds (V) and (VI) occurs. This side reaction complicates the product purification and reduces the yield, which is only approx. 58%, considering that the yield of compound (IV) prepared from (XIII) according to the state of the art is 72%. - [0007]
The method proposed in United States Patent 5,382,669 and Syn. Lett. 1995, 605-606 showed that catalysis by copper salts in the presence of phosphine ligands permitted the ethyl isocyanate reaction to be run at room temperature with only 3 equivalents of ethyl isocyanate. However, despite of moderation in reaction conditions the conversion and the ratio of cabergoline (I) and the byproducts (V and VI) are not much different from the uncatalyzed thermal reaction. The yield is only approx. 48% and 57%, considering that the yield of compound (IV) prepared from (XIII) according to the state of the art is 72%. - [0008]
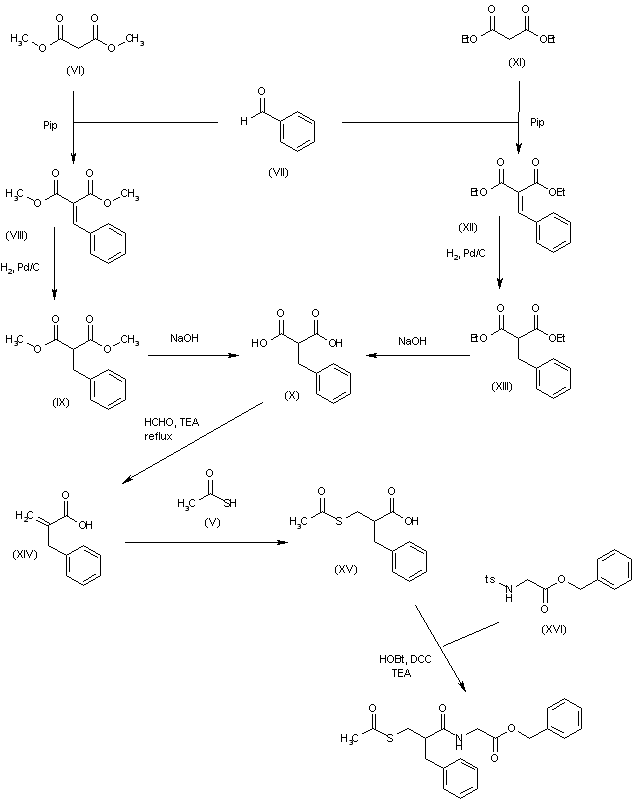
J. Org. Chem. 2002, 67, 7147-7150 describes an ethyl isocyanate-free method for the production of cabergoline (I) that solves the problem of completing acylation of indole nitrogen, too. - [0009]
The first step is the protection of indole nitrogen of amide (IV) preferably as tert-butyl carbamate (VII). - [0010]
Extension of the amide side chain is done by deprotonation of compound (VII) with sodium hexamethyldisilazide (NaHMDS) followed by trapping the anion with phenyl chloroformate (PhOCOCl) to yield the phenyl carbamate (VTII). - [0011]
Reaction of compound (VII) with ethylamine hydrochloride (EtNH2xHCl) gives BOC-cabergoline (IX) but also generates the ethylamide (X). The deprotection is done from the mixture of (IX) and (X) with 1N aqueous hydrochloric acid. The purified cabergoline (I) is then isolated by basification followed by chromatography on silica. (Scheme 3). - [0012]
In this approach the deprotonating step requires special cold reactor and strictly anhydrous circumstances. These requirements can hardly be satisfied in the course of large-scale preparation and the yield is only approx. 52%, considering that the yield of compound (VII) prepared from (XIII) according to the state of the art is 66%. - [0013]
According to US 2002/0177709 A1 Patent Application cabergoline (I) may be prepared by silylating amide (IV) with a silylating agent (e.g. trimethylsilyl trifluoromethane sulfonate – TMSOTf), reacting the obtained product (XI) with ethyl isocyanate (EtNCO) followed by desilylation of intermediate (XII) (Scheme 4). - [0014]
The disadvantage of this process is, that the silylating step requires strictly anhydrous circumstances. Otherwise, the reaction with ethyl isocyanate runs too long (24 hours) raising the safety hazard in the course of large-scale preparation and the yield is approx. 65%, considering that the yield of compound (IV) prepared from (XIII) according to the state of the art is 72%. - [0015]
Several crystalline forms of Cabergoline (I) are known. - [0016]
IL Farmaco 1995, 50 (3), 175-178 describes the preparation of crystalline form I. This solvated anhydrate product is crystallized from diethyl ether. - [0017]
WO 01/70740 A1 Patent Application describes a new process for the preparation of crystalline form I from the new crystalline form V. The form V – which is toluene solvate – is prepared from the mixture of the purified cabergoline (I) with toluene and diethyl ether by a long-lasting complicated process, at low reaction temperature, and the yield is only 45%. The crystalline form I is prepared by drying the form V in vacuum. - [0018]
WO 01/72746-A1 Patent Application describes the preparation of crystalline form VII from the crystalline form L By this process the suspension of form I in n-heptane or 1,4-dioxane is stirred for 48 hours, and then the suspension was filtered to obtain the crystalline form VII. The yield is 45.2%. - [0019]
WO 01/72747 A1 Patent Application describes the crystalline form II and a process for its preparation with approx. 70% yield by stirring the cabergoline (I) for several days in an organic solvent (eg. diethyl ether) at low temperature. - [0020]
Xenobiotica 1993, 23(12), 1377-1389, describes a comparison of the disposition and urinary metabolic pattern of 14C-cabergoline after single oral administration to rat, monkey and man. Among the potential metabolites of cabergoline a compound identified as FCE 27392 is disclosed, which corresponds to intermediate XIX of the preparation process of cabergoline according to the present invention.
The reaction procedure is shown in Scheme 5.

EXAMPLE 5Synthesis of 6-allyl-N-[3-(dimethylamino)propyl]-N-[(ethylamino)carbonyl]-ergoline-8β-carboxamide (I) (Cabergoline).
- [0077]
To a suspension of 9.0 g (21.87 mmol) N-[3-(dimethylamino)propyl]-N-[(ethylamino)carbonyl)-ergoline-8β-carboxamide (XIX) in 250 ml of toluene 0.5 g of tetrakis(triphenylphosphine)palladium(0) and 5 ml of allyl acetate was added, and the reaction mixture was stirred at ambient temperature for 2 hours. The resulting mixture was washed with 100 ml of water. The organic layer was dried over anhydrous sodium sulphate. The dried solution was concentrated in vacuum and the product was purified on a silica plug to give 9.1 g (92.3%) of the title compound.
1H NMR (DMSO-d6, TMS, 500MHz) δ 1.10 (t, 3H, J=7.2Hz, CONHCH2CH 3); 1.47 (q, 1H, J=12.4Hz, Hβ-9); 1.62-1.72 (m, 2H, CONCH2CH 2CH2N(CH3)2); 2.15 (s, 6H, CONCH2CH2CH2N(CH 3)2); 2.20-2.30 (m, 2H, CONHCH2CH2CH 2N(CH3)2); 2.32-2.40 (m, 2H, H-5, Hβ-7); 2.54 (dd, 1H, J=14.3Hz, 11.2Hz, Hα-4); 2.68-2.84 (m, 2H, Hα-9, H-10); 3.08 (ddd, 1H, J=11.3Hz, 3.1Hz, 1.8Hz, Hα-7); 3.14-3.22 (m, 2H, CONHCH 2CH3); 3.26 (dd, 1H, J=14.7Hz, 7.3Hz, Hx-N(6)CH 2CH=CH2); 3.28-3.38 (m, 2H, Hβ-4, H-8); 3.48 (dd, 1H, J=14.7Hz, 5.8Hz, Hy-N(6)CH 2CH=CH2); 3.58-3.68 (m, 2H, CONCH 2CH2CH2N(CH3)2); 5.15 (d, 1H, J=10.3Hz, Hx-N(6)CH2CH=CH 2); 5.24 (d, 1H, J=17.2Hz, Hy-N(6)CH2CH=CH 2); 5.88-5.98 (m, 1H, N(6)CH2CH=CH2); 6.75 (d, 1H, J=7.0Hz, H-12); 6.97 (s, 1H, H-2); 7.01 (t, 1H, J=7.5Hz, H-13); 7.13 (d, 1H, J=8.0Hz, H-14); 9.04 (t, 1H, J=5.0Hz, CONHCH2CH3); 10.60 (s, 1H, N(1)H).
EXAMPLE 6Production of amorphous form of Cabergoline (I)
- [0078]
- a) 10 g of chromatographically purified oily Cabergoline (I) was dissolved in 50 ml of acetone. The solution was concentrated in vacuum at 25-30°C to approx. 15 g. The obtained oily residue was dissolved in 40 ml of acetone, and the solution was concentrated in vacuum at 25-30°C to approx. 12 g. The obtained oily residue was dissolved in 30 ml of acetone again, and the solution was concentrated in vacuum at 25-30°C to 10 g. The obtained solid Cabergoline (I) was dried in vacuum at 25-30°C to solvent-free, to give 9.8 g (98%) of the title compound.
- b) The same as in Example 6a, but employing methyl acetate as solvent, 9.85 g (98.5%) of the title compound was obtained.
- c) The same as in Example 6a, but employing dichloromethane as solvent, 9.82 g (982%) of the title compound was obtained.
Patent
Publication numberPriority datePublication dateAssigneeTitleFamily To Family CitationsGB9205439D0 *1992-03-121992-04-22Erba Carlo SpaProcess for the synthesis of ergoline derivativesUS6696568B2 *2001-04-162004-02-24Finetech Ltd.Process and intermediates for production of cabergoline and related compoundsJP2004525187A *2001-04-162004-08-19フイネテク・リミテツドMethods and intermediates for the preparation of cabergoline and related compounds
Publication numberPriority datePublication dateAssigneeTitleFamily To Family CitationsEP1620101A4 *2003-05-082008-07-09Ivax Pharmaceuticals SroPolymorphs of cabergolineEP1925616A1 *2006-10-262008-05-28LEK Pharmaceuticals D.D.Process for the preparation of crystal forms of cabergoline via stable solvates of cabergolineWO2008104956A2 *2007-02-282008-09-04Ranbaxy Laboratories LimitedProcess for the preparation of amorphous cabergolineEP2083008A1 *2007-12-072009-07-29Axxonis Pharma AGErgoline derivatives as selective radical scavengers for neuronsWO2011154827A2 *2010-06-112011-12-15Rhodes TechnologiesTransition metal-catalyzed processes for the preparation of n-allyl compounds and use thereofCA2876321A1 *2012-06-222013-12-27Map Pharmaceuticals, Inc.Novel cabergoline derivatives
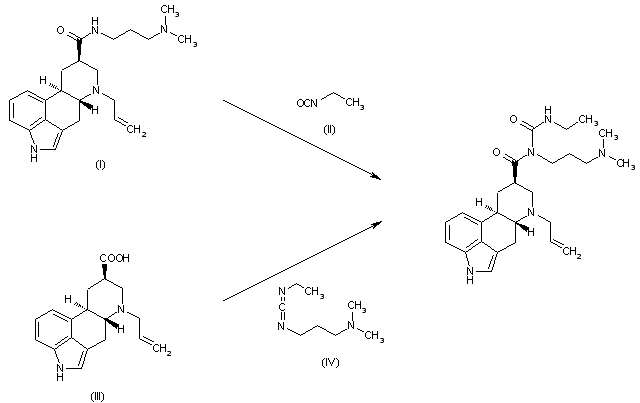
SYNBE 0894060; JP 58038282This compound can be obtained by two differents ways: 1) By condensation of 6-allyl-N-[3-(dimethylamino)propyl]ergolin-8-beta-caboxamide (I) with ethyl isocyanate (II) in refluxing toluene. 2) By condensation of 6-allylergolin-8-beta-carboxylic acid (III) with N-ethyl-N’-[3-(dimethylamino)propyl]carbodiimide (IV) in refluxing THF.
SYN
| J Label Compd Radiopharm 1991,29(5),519 |
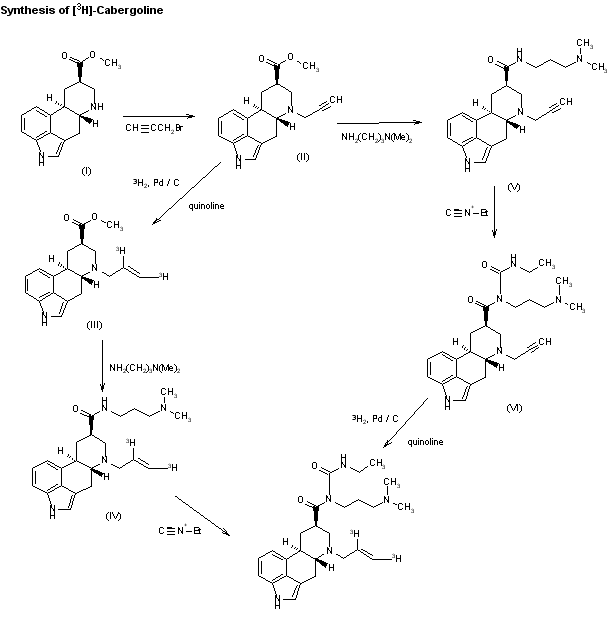
The synthesis of tritiated cabergoline by two similar routes has been described: 1) The acylation of 6-nor-dihydrolysergic acid methyl ester (I) with propargyl bromide yields the corresponding 6-propargyl derivative (II), which is hydrogenated with tritium gas over Pd/C in the presence of quinoline to give the ditritiated 6-allyl derivative (III). This compound is treated with 3-(dimethylamino)propylamine at 120 C, yielding the amide (IV), which is finally treated with ethyl isocyanate. 2) The reaction of the propargyl derivative (II) with 3-(dimethylamino)propylamine as before gives the amide (V). The reaction of (V) with ethyl isocyanate gives compound (VI), which is then hydrogenated with tritium as before.
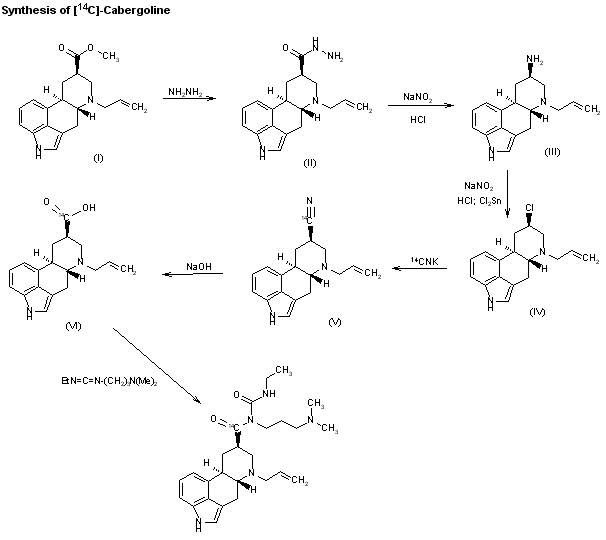
SYNThe synthesis of [14C]-cabergoline has also been described: The reaction of 6-allylergoline-8beta-carboxylic acid methyl ester (I) with hydrazine in refluxing methanol gives the hydrazide (II), which by reaction with NaNO2-HCl in water is converted to the amine (III). The reaction of (III) again with NaNO2-HCl in water, followed by reaction with SnCl2, affords the chloro derivative (IV), which is condensed with [14C]-CNK in refluxing ethanol-water yielding the nitrile (V). Hydrolysis of (V) with NaOH in refluxing ethanol affords the acid (VI), which is finally condensed with N-ethyl-N’-[3-(dimethylamino)propyl]carbodiimide in DMF.

AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT

join me on Linkedin
Anthony Melvin Crasto Ph.D – India | LinkedIn
join me on Researchgate
RESEARCHGATE

join me on Facebook
Anthony Melvin Crasto Dr. | Facebook
join me on twitter
Anthony Melvin Crasto Dr. | twitter
+919321316780 call whatsaapp
EMAIL. amcrasto@amcrasto
/////////////////////////////////////////////////////////////////////////////
Medical uses
- Lactation suppression
- Hyperprolactinemia[3]
- Adjunctive therapy of prolactin-producing pituitary gland tumors (prolactinomas);
- Monotherapy of Parkinson’s disease in the early phase;
- Combination therapy, together with levodopa and a decarboxylase inhibitor such as carbidopa, in progressive-phase Parkinson’s disease;
- In some countries also: ablactation and dysfunctions associated with hyperprolactinemia (amenorrhea, oligomenorrhea, anovulation, nonpuerperal mastitis and galactorrhea);
- Treatment of uterine fibroids.[4][5]
- Adjunctive therapy of acromegaly, cabergoline has low efficacy in suppressing growth hormone levels and is highly efficient in suppressing hyperprolactinemia that is present in 20-30% of acromegaly cases; growth hormone and prolactin are similar structurally and have similar effects in many target tissues, therefore targeting prolactin may help symptoms when growth hormone secretion can not be sufficiently controlled by other methods;
Cabergoline is frequently used as a first-line agent in the management of prolactinomas due to its higher affinity for D2 receptor sites, less severe side effects, and more convenient dosing schedule than the older bromocriptine, though in pregnancy bromocriptine is often still chosen since there is less data on safety in pregnancy for cabergoline.
Off-label
It has at times been used as an adjunct to SSRI antidepressants as there is some evidence that it counteracts certain side effects of those drugs, such as reduced libido and anorgasmia. It also has been suggested that it has a possible recreational use in reducing or eliminating the male refractory period, thereby allowing men to experience multiple ejaculatory orgasms in rapid succession, and at least two scientific studies support those speculations.[6][7]: e28–e33 Additionally, a systematic review and meta-analysis concluded that prophylactic treatment with cabergoline reduces the incidence, but not the severity, of ovarian hyperstimulation syndrome (OHSS), without compromising pregnancy outcomes, in females undergoing stimulated cycles of in vitro fertilization (IVF).[8] Also, a study on rats found that cabergoline reduces voluntary alcohol consumption, possibly by increasing GDNF expression in the ventral tegmental area.[9] It may be used in the treatment of restless legs syndrome.[citation needed]
Pregnancy and lactation
Relatively little is known about the effects of this medication during pregnancy and lactation. In some cases the related bromocriptine may be an alternative when pregnancy is expected.[citation needed]
- Pregnancy: available preliminary data indicates a somewhat increased rate of congenital abnormalities in patients who became pregnant while treated with cabergoline.[citation needed]. However, one study concluded that “foetal exposure to cabergoline through early pregnancy does not induce any increase in the risk of miscarriage or foetal malformation.”[10]
- Lactation: In rats cabergoline was found in the maternal milk. Since it is not known if this effect also occurs in humans, breastfeeding is usually not recommended if/when treatment with cabergoline is necessary.
- Lactation suppression: In some countries cabergoline (Dostinex) is sometimes used as a lactation suppressant. It is also used in veterinary medicine to treat false pregnancy in dogs.
Contraindications
- Hypersensitivity to ergot derivatives
- Pediatric patients (no clinical experience)
- Severely impaired liver function or cholestasis
- Concomitant use with drugs metabolized mainly by CYP450 enzymes such as erythromycin and ketoconazole, because increased plasma levels of cabergoline may result (although cabergoline undergoes minimal CYP450 metabolism).
- Cautions: severe cardiovascular disease, Raynaud’s disease, gastroduodenal ulcers, active gastrointestinal bleeding, hypotension.
Side effects
Side effects are mostly dose dependent. Much more severe side effects are reported for treatment of Parkinson’s disease and (off-label treatment) for restless leg syndrome which both typically require very high doses. The side effects are considered mild when used for treatment of hyperprolactinemia and other endocrine disorders or gynecologic indications where the typical dose is one hundredth to one tenth that for Parkinson’s disease.[citation needed]
Cabergoline requires slow dose titration (2–4 weeks for hyperprolactinemia, often much longer for other conditions) to minimise side effects. The extremely long bioavailability of the medication may complicate dosing regimens during titration and require particular precautions.
Cabergoline is considered the best tolerable option for hyperprolactinemia treatment although the newer and less tested quinagolide may offer similarly favourable side effect profile with quicker titration times.
Approximately 200 patients with newly diagnosed Parkinson’s disease participated in a clinical study of cabergoline monotherapy.[11] Seventy-six (76) percent reported at least one side effect. These side effects were chiefly mild or moderate:
- GI tract: Side effects were extremely frequent. Fifty-three percent of patients reported side effects. Very frequent: Nausea (30%), constipation (22%), and dry mouth (10%). Frequent: Gastric irritation (7%), vomiting (5%), and dyspepsia (2%).
- Psychiatric disturbances and central nervous system (CNS): Altogether 51 percent of patients were affected. Very frequent: Sleep disturbances (somnolence 18%, insomnia 11%), vertigo (27%), and depression (13%). Frequent: dyskinesia (4%) and hallucinations (4%).
- Cardiovascular: Approximately 30 percent of patients experienced side effects. Most frequent were hypotension (10%), peripheral edema (14%) and non-specific edema (2%). Arrhythmias were encountered in 4.8%, palpitations in 4.3%, and angina pectoris in 1.4%.
In a combination study with 2,000 patients also treated with levodopa, the incidence and severity of side effects was comparable to monotherapy. Encountered side effects required a termination of cabergoline treatment in 15% of patients. Additional side effects were infrequent cases of hematological side effects, and an occasional increase in liver enzymes or serum creatinine without signs or symptoms.
As with other ergot derivatives, pleuritis, exudative pleura disease, pleura fibrosis, lung fibrosis, and pericarditis are seen. These side effects are noted in less than 2% of patients. They require immediate termination of treatment. Clinical improvement and normalization of X-ray findings are normally seen soon after cabergoline withdrawal. It appears that the dose typically used for treatment of hyperprolactinemia is too low to cause this type of side effects.
Valvular heart disease
In two studies published in the New England Journal of Medicine on January 4, 2007, cabergoline was implicated along with pergolide in causing valvular heart disease.[12][13] As a result of this, the FDA removed pergolide from the U.S. market on March 29, 2007.[14] Since cabergoline is not approved in the U.S. for Parkinson’s Disease, but for hyperprolactinemia, the drug remains on the market. The lower doses required for treatment of hyperprolactinemia have been found to be not associated with clinically significant valvular heart disease or cardiac valve regurgitation.[15][16]
Interactions
No interactions were noted with levodopa or selegiline. The drug should not be combined with other ergot derivatives. Dopamine antagonists such as antipsychotics and metoclopramide counteract some effects of cabergoline. The use of antihypertensive drugs should be intensively monitored because excessive hypotension may result from the combination.
Pharmacology
Pharmacodynamics
| Site | Affinity (Ki [nM]) | Efficacy (Emax [%]) | Action |
|---|---|---|---|
| D1 | 214–32,000 | ? | ? |
| D2S | 0.5–0.62 | 102 | Full agonist |
| D2L | 0.95 | 75 | Partial agonist |
| D3 | 0.80–1.0 | 86 | Partial agonist |
| D4 | 56 | 49 | Partial agonist |
| D5 | 22 | ? | ? |
| 5-HT1A | 1.9–20 | 93 | Partial agonist |
| 5-HT1B | 479 | 102 | Full agonist |
| 5-HT1D | 8.7 | 68 | Partial agonist |
| 5-HT2A | 4.6–6.2 | 94 | Partial agonist |
| 5-HT2B | 1.2–9.4 | 123 | Full agonist |
| 5-HT2C | 5.8–692 | 96 | Partial agonist |
| 5-HT3 | >10,000 | – | – |
| 5-HT4 | 3,000 | ? | ? |
| 5-HT6 | 1,300 | ? | ? |
| 5-HT7 | 2.5 | ? | Antagonist |
| α1A | 288–>10,000 | 0 | Silent antagonist |
| α1B | 60–1,000 | ? | ? |
| α1D | 166 | ? | ? |
| α2A | 12–132 | 0 | Silent antagonist |
| α2B | 17–72 | 0 | Silent antagonist |
| α2C | 22–364 | 0 | Silent antagonist |
| α2D | 3.6 | ? | ? |
| H1 | 1,380 | ? | ? |
| M1 | >10,000 | – | – |
| SERT | >10,000 | – | – |
| Notes: All sites are human except α2D-adrenergic, which is rat (no human counterpart).[17] Negligible affinity (>10,000 nM) for various other receptors (β1– and β2-adrenergic, adenosine, GABA, glutamate, glycine, nicotinic acetylcholine, opioid, prostanoid).[18] Sources: [17][19][20][18][21] |
Cabergoline is a long-acting dopamine D2 receptor agonist. In-vitro rat studies show a direct inhibitory effect of cabergoline on the prolactin secretion in the lactotroph cells of the pituitary gland and cabergoline decreases serum prolactin levels in reserpinized rats.[citation needed] Although cabergoline is commonly described principally as a D2 receptor agonist, it also possesses significant affinity for the dopamine D3, and D4, serotonin 5-HT1A, 5-HT2A, 5-HT2B, and 5-HT2C, and α2-adrenergic receptors, as well as moderate/low affinity for the dopamine D1, serotonin 5-HT7, and α1-adrenergic receptors.[17][18][22] Cabergoline functions as an partial or full agonist at all of these receptors except for the 5-HT7, α1-adrenergic, and α2-adrenergic receptors, where it acts as an antagonist.[19][20][18] Cabergoline has been associated with cardiac valvulopathy due to activation of 5-HT2B receptors.[23]
Pharmacokinetics
Following a single oral dose, resorption of cabergoline from the gastrointestinal (GI) tract is highly variable, typically occurring within 0.5 to 4 hours. Ingestion with food does not alter its absorption rate. Human bioavailability has not been determined since the drug is intended for oral use only. In mice and rats the absolute bioavailability has been determined to be 30 and 63 percent, respectively. Cabergoline is rapidly and extensively metabolized in the liver and excreted in bile and to a lesser extent in urine. All metabolites are less active than the parental drug or inactive altogether. The human elimination half-life is estimated to be 63 to 68 hours in patients with Parkinson’s disease and 79 to 115 hours in patients with pituitary tumors. Average elimination half-life is 80 hours.
The therapeutic effect in treatment of hyperprolactinemia will typically persist for at least 4 weeks after cessation of treatment.
History
Cabergoline was first synthesized by scientists working for the Italian drug company Farmitalia-Carlo Erba in Milan who were experimenting with semisynthetic derivatives of the ergot alkaloids, and a patent application was filed in 1980.[24][25][26] The first publication was a scientific abstract at the Society for Neuroscience meeting in 1991.[27][28]
Farmitalia-Carlo Erba was acquired by Pharmacia in 1993,[29] which in turn was acquired by Pfizer in 2003.[30]
Cabergoline was first marketed in The Netherlands as Dostinex in 1992.[24] The drug was approved by the FDA on December 23, 1996.[31] It went generic in late 2005 following US patent expiration.[32]
Society and culture
Brand names
Brand names of cabergoline include Cabaser, Dostinex, Galastop (veterinary), and Kelactin (veterinary), among others.[33]
Research
Cabergoline was studied in one person with Cushing’s disease, to lower adrenocorticotropic hormone (ACTH) levels and cause regression of ACTH-producing pituitary adenomas.[34]
References
- ^ J. Elks; C. R. Ganellin (1990). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 204–.
- ^ Fischer, Jnos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 533. ISBN 9783527607495.
- ^ UK electronic Medicines Compendium Dostinex Tablets Last Updated on eMC Dec 23, 2013
- ^ Sayyah-Melli, M; Tehrani-Gadim, S; Dastranj-Tabrizi, A; Gatrehsamani, F; Morteza, G; Ouladesahebmadarek, E; Farzadi, L; Kazemi-Shishvan, M (2009). “Comparison of the effect of gonadotropin-releasing hormone agonist and dopamine receptor agonist on uterine myoma growth. Histologic, sonographic, and intra-operative changes”. Saudi Medical Journal. 30 (8): 1024–33. PMID 19668882.
- ^ Sankaran, S.; Manyonda, I. (2008). “Medical management of fibroids”. Best Practice & Research Clinical Obstetrics & Gynaecology. 22 (4): 655–76. doi:10.1016/j.bpobgyn.2008.03.001. PMID 18468953. http://www.britishfibroidtrust.org.uk/journals/bft_Sankaran.pdf
- ^ Krüger TH, Haake P, Haverkamp J, et al. (December 2003). “Effects of acute prolactin manipulation on sexual drive and function in males”. Journal of Endocrinology. 179 (3): 357–65. CiteSeerX 10.1.1.484.4005. doi:10.1677/joe.0.1790357. PMID 14656205.
- ^ Hollander, Adam B.; Pastuszak, Alexander W.; Lipshultz, Larry I. (2016). “Cabergoline in the Treatment of Male Orgasmic Disorder—A Retrospective Pilot Analysis”. Journal of Sexual Medicine. 4 (4): e28–e33. doi:10.1016/j.esxm.2015.09.001. PMC 4822480. PMID 26944776.
- ^ Youssef MA, van Wely M, Hassan MA, et al. (March 2010). “Can dopamine agonists reduce the incidence and severity of OHSS in IVF/ICSI treatment cycles? A systematic review and meta-analysis”. Hum Reprod Update. 16 (5): 459–66. doi:10.1093/humupd/dmq006. PMID 20354100.
- ^ Carnicella, S.; Ahmadiantehrani, S.; He, D. Y.; Nielsen, C. K.; Bartlett, S. E.; Janak, P. H.; Ron, D. (2009). “Cabergoline Decreases Alcohol Drinking and Seeking Behaviors Via Glial Cell Line-Derived Neurotrophic Factor”. Biological Psychiatry. 66 (2): 146–153. doi:10.1016/j.biopsych.2008.12.022. PMC 2895406. PMID 19232578.
- ^ Colao, A; Abs R.; et al. (January 2008). “Pregnancy outcomes following cabergoline treatment: extended results from a 12-year observational study”. Clinical Endocrinology. 68 (1): 66–71. doi:10.1111/j.1365-2265.2007.03000.x. PMID 17760883. S2CID 38408935.
- ^ Rinne, U. K.; Bracco, F.; Chouza, C.; Dupont, E.; Gershanik, O.; Masso, J. F. M.; Montastruc, J. L.; Marsden, C. D.; Dubini, A.; Orlando, N.; Grimaldi, R. (1997-02-01). “Cabergoline in the treatment of early parkinson’s disease: Results of the first year of treatment in a double-blind comparison of cabergoline and levodopa”. Neurology. 48 (2): 363–368. doi:10.1212/WNL.48.2.363. ISSN 0028-3878. PMID 9040722. S2CID 34955541.
- ^ Schade, Rene; Andersohn, Frank; Suissa, Samy; Haverkamp, Wilhelm; Garbe, Edeltraut (2007-01-04). “Dopamine Agonists and the Risk of Cardiac-Valve Regurgitation”. New England Journal of Medicine. 356 (1): 29–38. doi:10.1056/NEJMoa062222. PMID 17202453.
- ^ Zanettini, Renzo; Antonini, Angelo; Gatto, Gemma; Gentile, Rosa; Tesei, Silvana; Pezzoli, Gianna (2007-01-04). “Valvular Heart Disease and the Use of Dopamine Agonists for Parkinson’s Disease”. New England Journal of Medicine. 356 (1): 39–46. doi:10.1056/NEJMoa054830. PMID 17202454.
- ^ “Food and Drug Administration Public Health Advisory”. Food and Drug Administration. 2007-03-29. Archived from the original on 2007-04-08. Retrieved 2007-04-27.
- ^ Bogazzi, F.; Buralli, S.; Manetti, L.; Raffaelli, V.; Cigni, T.; Lombardi, M.; Boresi, F.; Taddei, S.; Salvetti, A. (2008). “Treatment with low doses of cabergoline is not associated with increased prevalence of cardiac valve regurgitation in patients with hyperprolactinaemia”. International Journal of Clinical Practice. 62 (12): 1864–9. doi:10.1111/j.1742-1241.2008.01779.x. PMID 18462372. S2CID 7822137.
- ^ Wakil, A.; Rigby, A. S; Clark, A. L; Kallvikbacka-Bennett, A.; Atkin, S. L (2008). “Low dose cabergoline for hyperprolactinaemia is not associated with clinically significant valvular heart disease”. European Journal of Endocrinology. 159 (4): R11–4. doi:10.1530/EJE-08-0365. PMID 18625690.
- ^ Jump up to:a b c Millan MJ, Maiofiss L, Cussac D, Audinot V, Boutin JA, Newman-Tancredi A (November 2002). “Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes”. J Pharmacol Exp Ther. 303 (2): 791–804. doi:10.1124/jpet.102.039867. PMID 12388666. S2CID 6200455.
- ^ Jump up to:a b c d Sharif NA, McLaughlin MA, Kelly CR, Katoli P, Drace C, Husain S, Crosson C, Toris C, Zhan GL, Camras C (March 2009). “Cabergoline: Pharmacology, ocular hypotensive studies in multiple species, and aqueous humor dynamic modulation in the Cynomolgus monkey eyes”. Experimental Eye Research. 88 (3): 386–97. doi:10.1016/j.exer.2008.10.003. PMID 18992242.
- ^ Jump up to:a b Newman-Tancredi A, Cussac D, Audinot V, Nicolas JP, De Ceuninck F, Boutin JA, Millan MJ (November 2002). “Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. II. Agonist and antagonist properties at subtypes of dopamine D(2)-like receptor and alpha(1)/alpha(2)-adrenoceptor”. J Pharmacol Exp Ther. 303 (2): 805–14. doi:10.1124/jpet.102.039875. PMID 12388667. S2CID 35238120.
- ^ Jump up to:a b Newman-Tancredi A, Cussac D, Quentric Y, Touzard M, Verrièle L, Carpentier N, Millan MJ (November 2002). “Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. III. Agonist and antagonist properties at serotonin, 5-HT(1) and 5-HT(2), receptor subtypes”. J Pharmacol Exp Ther. 303 (2): 815–22. doi:10.1124/jpet.102.039883. PMID 12388668. S2CID 19260572.
- ^ https://web.archive.org/web/20210413033753/https://pdsp.unc.edu/databases/pdsp.php?testFreeRadio=testFreeRadio&testLigand=Cabergoline&doQuery=Submit+Query
- ^ National Institute of Mental Health. PDSD Ki Database (Internet) [cited 2013 Jul 24]. ChapelHill (NC): University of North Carolina. 1998-2013. Available from: “Archived copy”. Archived from the original on 2013-11-08. Retrieved 2014-03-04.
- ^ Cavero I, Guillon JM (2014). “Safety Pharmacology assessment of drugs with biased 5-HT(2B) receptor agonism mediating cardiac valvulopathy”. J Pharmacol Toxicol Methods. 69 (2): 150–61. doi:10.1016/j.vascn.2013.12.004. PMID 24361689.
- ^ Jump up to:a b Council regulation (EEC) no 1768/92 in the matter of Application No SPC/GB94/012 for a Supplementary Protection Certificate in the name of Farmitalia Carlo Erba S. r. l.
- ^ Espace record: GB 202074566
- ^ US Patent 4526892 – Dimethylaminoalkyl-3-(ergoline-8′.beta.carbonyl)-ureas
- ^ Fariello, RG (1998). “Pharmacodynamic and pharmacokinetic features of cabergoline. Rationale for use in Parkinson’s disease”. Drugs. 55 (Suppl 1): 10–6. doi:10.2165/00003495-199855001-00002. PMID 9483165. S2CID 46973281.
- ^ Carfagna N, Caccia C, Buonamici M, Cervini MA, Cavanus S, Fornaretto MG, Damiani D, Fariello RG (1991). “Biochemical and pharmacological studies on cabergoline, a new putative antiparkinsonian drug”. Soc Neurosci Abs. 17: 1075.
- ^ Staff. News: Farmitalia bought by Kabi Pharmacia[permanent dead link]. Ann Oncol (1993) 4 (5): 345.
- ^ Staff, CNN/Money. April 16, 2003 It’s official: Pfizer buys Pharmacia
- ^ FDA approval history
- ^ “Drugs@FDA: FDA Approved Drug Products – ANDA 076310”. http://www.accessdata.fda.gov. FDA.gov. Retrieved 14 December 2018.
- ^ “Cabergoline Uses, Side Effects & Warnings”. Archived from the original on 2015-12-30.
- ^ Miyoshi, T.; et al. (2004). “Effect of cabergoline treatment on Cushing’s disease caused by aberrant adrenocorticotropin-secreting macroadenoma”. Journal of Endocrinological Investigation. 27 (11): 1055–1059. doi:10.1007/bf03345309. PMID 15754738. S2CID 6660262.
External links
| Clinical data | |
|---|---|
| Trade names | Dostinex, others |
| AHFS/Drugs.com | Monograph |
| License data | US FDA: CABERGOLINE |
| Routes of administration | Oral |
| ATC code | G02CB03 (WHO) N04BC06 (WHO) |
| Legal status | |
| Legal status | US: ℞-only |
| Pharmacokinetic data | |
| Bioavailability | First-pass effect seen; absolute bioavailability unknown |
| Protein binding | Moderately bound (40–42%); concentration-independent |
| Metabolism | Hepatic, predominately via hydrolysis of the acylurea bond or the urea moiety |
| Elimination half-life | 63–69 hours (estimated) |
| Excretion | Urine (22%), feces (60%) |
| Identifiers | |
| showIUPAC name | |
| CAS Number | 81409-90-7 |
| PubChem CID | 54746 |
| IUPHAR/BPS | 37 |
| DrugBank | DB00248 |
| ChemSpider | 49452 |
| UNII | LL60K9J05T |
| KEGG | D00987 |
| ChEBI | CHEBI:3286 |
| ChEMBL | ChEMBL1201087 |
| CompTox Dashboard (EPA) | DTXSID6022719 |
| ECHA InfoCard | 100.155.380 |
| Chemical and physical data | |
| Formula | C26H37N5O2 |
| Molar mass | 451.615 g·mol−1 |
| 3D model (JSmol) | Interactive image |
| showSMILES | |
| showInChI | |
| (what is this?) (verify) |
////////////////Cabergoline, UNII-LL60K9J05T, каберголин , كابارغولين ,卡麦角林 , FCE-21336
[H][C@@]12CC3=CNC4=CC=CC(=C34)[C@@]1([H])C[C@H](CN2CC=C)C(=O)N(CCCN(C)C)C(=O)NCC


 All my awards,
All my awards, 










































































































































![{\displaystyle [\alpha ]_{D}^{20}}](http://wikimedia.org/api/rest_v1/media/math/render/svg/b67b300e0d318e964fcdab92c872ed89d1d4f43f) +65° to +72° cm³/dm·g (1% in
+65° to +72° cm³/dm·g (1% in