sodium;1,4-bis(2-ethylhexoxy)-1,4-dioxobutane-2-sulfonate CAS Registry Number: 577-11-7 CAS Name: Sulfobutanedioic acid 1,4-bis(2-ethylhexyl) ester sodium salt Additional Names: sulfosuccinic acid 1,4-bis(2-ethylhexyl) ester S-sodium salt; bis(2-ethylhexyl)sodium sulfosuccinate; dioctyl sodium sulfosuccinate; sodium dioctyl sulfosuccinate; DSS Trademarks: Aerosol OT (Cyanamid); Colace (Roberts); Comfolax (Searle); Coprola (Dunster); Dioctylal (Continental Pharma); Dioctyl (Medo); Diotilan (Chinoin); Disonate (Lannett); Doxinate (Hoechst); Doxol (Blair); Dulcivac (Harvey); Jamylène (Thaplix); Molatoc; Molcer (Wallace); Nevax; Regutol (Schering-Plough); Soliwax (Concept Pharm.); Velmol (Berlex); Waxsol (Norgine); Yal (Ritter) Molecular Formula: C20H37NaO7S Molecular Weight: 444.56 Percent Composition: C 54.03%, H 8.39%, Na 5.17%, O 25.19%, S 7.21% Literature References: Prepn: Jaeger, US2028091; US2176423 (1936, 1939, both to Am. Cyanamid). Structure and wetting power: Caryl, Ind. Eng. Chem.33, 731 (1941). Comprehensive description: S. Ahuja, J. Cohen, Anal. Profiles Drug Subs.2, 199-219 (1973); 12, 713-720 (1983). For structure see Docusate calcium. Properties: Available as wax-like solid, usually in rolls of tissue-thin material; also as 50-75% solns in various solvents. Soly in water (g/l): 15 (25°), 23 (40°), 30 (50°), 55 (70°). Sol in CCl4, petr ether, naphtha, xylene, dibutyl phthalate, liq petrolatum, acetone, alcohol, vegetable oils. Very sol in water + alcohol, water + water-miscible organic solvents. Stable in acid and neutral solns; hydrolyzes in alkaline solns. Derivative Type: Docusate potassium CAS Registry Number: 7491-09-0 Trademarks: Rectalad (Carter-Wallace) Molecular Formula: C20H37KO7S Molecular Weight: 460.67 Percent Composition: C 52.14%, H 8.10%, K 8.49%, O 24.31%, S 6.96% NOTE: Ingredient of the laxative Peri-Colace (Roberts) which also contains casanthranol.Use: Sodium salt as pharmaceutic aid (surfactant); as wetting agent in industrial, pharmaceutical, cosmetic and food applications; dispersing and solubilizing agent in foods; adjuvant in tablet formation. Therap-Cat: Stool softener. Therap-Cat-Vet: Stool softener. Keywords: Laxative/Cathartic.
Docusate Calcium CAS Registry Number: 128-49-4 CAS Name: Sulfobutanedioic acid 1,4-bis(2-ethylhexyl)ester calcium salt Additional Names: bis[2-ethylhexyl]calcium sulfosuccinate; calcium dioctyl sulfosuccinate; dioctyl calcium sulfosuccinate Trademarks: Surfak (HMR) Molecular Formula: C40H74CaO14S2 Molecular Weight: 883.22 Percent Composition: C 54.40%, H 8.44%, Ca 4.54%, O 25.36%, S 7.26% Literature References: Prepd from dioctyl sodium sulfosuccinate dissolved in isopropanol and from calcium chloride dissolved in methanol: Klotz, US3035973 (1962 to Lloyd Brothers). Properties: White precipitate. Sol in mineral and vegetable oils, liq polyethylene glycol. Practically insol in glycerol. Claimed to have greater surface-active wetting properties than the sodium salt. NOTE: Ingredient of Doxidan (HMR) which also contains phenolphthalein. Therap-Cat: Stool softener. Keywords: Laxative/Cathartic.
Synthesis of Trihexyltetradecylphosphonium octylsulfosuccinate [P6, 6, 6, 14][docusate]
SYN
Docusate is the common chemical and pharmaceutical name of the anionbis(2-ethylhexyl) sulfosuccinate, also commonly called dioctyl sulfosuccinate (DOSS).[2][3][4]
Sodium docusate was patented in 1937 by Coleman R. Caryl and Alphons O. Jaeger for American Cyanamid,[3] which commercialized it for many years as a detergent under the brand name Aerosol OT.
Its use for the treatment of constipation was first proposed in 1955 by James L. Wilson and David G. Dickinson,[4] and quicky popularized under the name Doxinate.[13]
Medical use
Constipation
The main medical use of docusate sodium is to treat constipation, acting as a laxative and stool softener. In painful anorectal conditions such as hemorrhoid and anal fissures, it can help avoid pain caused by straining during bowel movements.
When administered by mouth, a bowel movement often occurs in 1 to 3 days,[1] while rectal use may be effective within 20 minutes.[14]
Sodium docusate is recommended as a stool softener for children.[1]
However, its effectiveness for constipation is poorly supported by evidence.[7][8] Multiple studies have found docusate to be no more effective than a placebo for improving constipation.[7][8][9][10] Others have found it to be less useful for the treatment of chronic constipation than psyllium.[10][15][16]
The medication may be given to people who are receiving opioid medication, although prolonged use may cause irritation of the gastrointestinal tract.[10][16]
Other medical uses
Docusate sodium, when used with ear syringing, may help with earwax removal, particularly in the case of impaction.[17]
When taken by mouth it should be ingested with plenty of water.
Side effects
Side effects are uncommon and typically mild,[1] and may include stomach pain, abdominal cramps or diarrhea,[1] Efficacy decreases with long-term use, and may cause poor bowel function.[11]
Serious allergic reactions may occur with the drug. The most severe side effect of docusate, although very rare, is rectal bleeding.[21]
Interactions
Docusate might increase resorption of other drugs, for example, dantron (1,8-dihydroxyanthraquinone).[16]
Mechanism of action
Docusate sodium works by allowing more water to be absorbed by the stool.[11][22]
Docusate does not stay in the gastrointestinal tract, but is absorbed into the bloodstream and excreted via the gallbladder[16] after undergoing extensive metabolism.
The effect of docusate may not necessarily be all due to its surfactant properties. Perfusion studies suggest that docusate inhibits fluid absorption or stimulates secretion in the portion of the small intestine known as the jejunum.
Pharmaceutical brand names
In the U.S., docusate sodium for pharmaceutical use is available under multiple brand names: Aqualax, Calube, Colace, Colace Micro-Enema, Correctol Softgel Extra Gentle, DC-240, Dialose, Diocto, Dioctocal, Dioctosoftez, Dioctyn, Dionex, Doc-Q-Lace, Docu Soft, Docucal, Doculax, Docusoft S, DOK, DOS, Doss-Relief, DSS, Dulcolax – Stool Softener (not to be confused with another drug marketed under the Dulcolax brand, bisacodyl, which is a stimulant laxative), Ex-Lax Stool Softener, Fleet Sof-Lax, Genasoft, Kasof, Laxa-basic, Modane Soft, Octycine-100, Pedia-Lax, Preferred Plus Pharmacy Stool Softener, Regulax SS, Sulfalax Calcium, Sur-Q-Lax, Surfak Stool Softener, and Therevac-SB. Generic preparations are also available.
In the UK, dioctyl sodium sulfosuccinate is sold under the brand name Docusol (Typharm Ltd) and DulcoEase (Boehringer Ingelheim).
In Australia, dioctyl sodium sulfosuccinate is sold as Coloxyl and Coloxyl with senna.
In India, preparations include Laxatin by Alembic, Doslax by Raptakos Laboratories, Cellubril by AstraZeneca, and Laxicon by Stadmed.
Other uses
Dioctyl sodium sulfosuccinate is used as a surfactant in a wide range of applications, often under the name Aerosol-OT.[4][23] It is unusual in that it is able to form microemulsions without the use of co-surfactants, and it has a rich variety of aqueous-phase behavior including multiple liquid crystalline phases.[24]
Food additive
Dioctyl sodium sulfosuccinate has been approved by the US FDA as a “generally recognized as safe” (GRAS) additive.[25] It is used in a variety of food products, as a surface active agent, stabilizer, thickener, wetting agent, processing aid, solubilizing agent, emulsifier, and dispersant. The highest amount found in food products is 0.5% by weight, which include pasteurized cheese spreads, cream cheeses and salad dressings.[26] The FDA also approved its use as a wetting agent or solubilizer for flavoring agents in carbonated and non-carbonated drinks at levels up to 10 parts per million.[25]
As a surfactant, docusate sodium is or has been commercialized under many brand names, including DSSj Aerosol OT, Alphasol OT, Colace, Complemix, Coprol, Dioctylal, Dioctyl-Medo Forte, Diotilan, Diovac, Disonate, Doxinate, Doxol, Dulsivac, Molatoc, Molofac, Nevax, Norval, Regutol, Softili, Solusol, Sulfimel DOS, Vatsol OT, Velmol, and Waxsol[28]
Ingestion may cause the side effects described above, such as diarrhea, intestinal bloating, and occasionally cramping pains. Dioctyl sodium sulfosuccinate is not known to be carcinogenic, mutagenic, or teratogenic.[29]
In a 2010 study, dioctyl sodium sulfosuccinate exhibited higher toxicity against bacteria (Vibrio fischeri, Anabaena sp.) and algae (Pseudokirchneriella subcapitata) than did a number of fluorinated surfactants (PFOS, PFOA, or PFBS). Measuring bioluminescence inhibition of the bacteria and growth inhibition of the algae, the LD50 were in the range of 43–75 mg/l. Combinations of the fluorinated compounds with dioctyl sodium sulfosuccinate showed mid to highly synergistic effects in most settings, meaning that such combinations are significantly more toxic than the individual substances.[30]
Freshwater species
The substance is highly toxic for rainbow trout with a median lethal concentration (LC50) of 0.56 mg/l after 48 hours for the pure substance. It is only slightly to moderately toxic for rainbow trout fingerlings, and slightly toxic for harlequin rasboras (LC50 27 mg/l of a 60% formulation after 48 hours).
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06.
^ Jump up to:abcd Ramkumar D, Rao SS (April 2005). “Efficacy and safety of traditional medical therapies for chronic constipation: systematic review”. The American Journal of Gastroenterology. 100 (4): 936–71. PMID15784043.
^ Friedman M (October 1956). “Dioctyl sodium sulfosuccinate (doxinate) in chronic functional constipation”. American Practitioner and Digest of Treatment. 7 (10): 1588–91. PMID13362832.
^ Mahadevan U, Kane S (July 2006). “American gastroenterological association institute medical position statement on the use of gastrointestinal medications in pregnancy”. Gastroenterology. 131(1): 278–82. doi:10.1053/j.gastro.2006.04.048. PMID16831610.
Hydroxypioglitazone is a member of the class of thiazolidenediones that is the hydroxy derivative of pioglitazone. It has a role as a human xenobiotic metabolite. It is a member of thiazolidinediones, a member of pyridines and an aromatic ether. It derives from a pioglitazone.
OriginatorIDIBELL
DeveloperMinoryx Therapeutics
ClassNeuroprotectants; Phenyl ethers; Pyridines; Small molecules; Thiazolidinediones
Mechanism of ActionPeroxisome proliferator-activated receptor gamma agonists
Orphan Drug StatusYes – Adrenoleucodystrophy; Friedreich’s ataxia
Phase II/IIIAdrenoleucodystrophy
Phase IIFriedreich’s ataxia
PreclinicalCNS disorders
23 Sep 2020Leriglitazone receives Rare Pediatric Disease designation from the US FDA for X-linked adrenoleukodystrophy before September 2020
23 Sep 2020Minoryx Therapeutics licenses leriglitazone to Sperogenix Therapeutics in China, Hong Kong and Macau for X-linked adrenoleukodystrophy (X-ALD)
14 Sep 2020Minoryx Therapeutics completes the phase II FRAMES trial in Friedreich’s ataxia (In adolescents, In adults) in Spain, Germany, France and Belgium (PO) (NCT03917225)
Leriglitazone (Hydroxypioglitazone), a metabolite of pioglitazone. Leriglitazone (Hydroxypioglitazone) PioOH is a PPARγ agonist, stabilizes the PPARγ activation function-2 (AF-2) co-activator binding surface and enhances co-activator binding, affording slightly better transcriptional efficacy. Leriglitazone (Hydroxypioglitazone) binds to the PPARγ C-terminal ligand-binding domain (LBD) with Ki of 1.2 μM,induces transcriptional efficacy of the PPARγ (LBD) with EC50 of 680 nM.
Leriglitazone is under investigation in clinical trial NCT03917225 (A Clinical Study to Evaluate the Effect of MIN-102 on the Progression of Friedreich’s Ataxia in Male and Female Patients).
Treatment of X-Linked Adrenoleukodystrophy
PATENT
WO 9218501
WO 9322445
PAPER
Chemical & Pharmaceutical Bulletin (1995), 43(12), 2168-72
The metabolites of (±)-5-[p-[2-(5-ethyl-2-pyridyl)ethoxy]benzyl]-2, 4-thiazolidinedione (1, pioglitazone), which is a representative insulin-sensitizing agent, were synthesized to confirm their structures and for studies of their pharmacological properties. Of the metabolites identified, a compound hydroxylated at the 2-position of the ethoxy chain (3) and compounds oxygenated at the ethyl side chain attached to the pyridine ring (4, 5) were found to be active, although the potency was slightly lower than that of the parent compound.
PAPER
Journal of Medicinal Chemistry (1996), 39(26), 5053-5063.
Pioglitazone (5-(4-(2-(5-ethyl-2-pyridyl)ethoxy)benzyl)-2,4-thiazolidinedione, 2) is a prototypical antidiabetic thiazolidinedione that had been evaluated for possible clinical development. Metabolites 6−9 have been identified after dosing of rats and dogs. Ketone 10 has not yet been identified as a metabolite but has been added to the list as a putative metabolite by analogy to alcohol 6 and ketone 7. We have developed improved syntheses of pioglitazone (2) metabolites 6−9 and the putative metabolite ketone 10. These entities have been compared in the KKAy mouse model of human type-II diabetes to pioglitazone (2). Ketone 10 has proven to be the most potent of these thiazolidinediones in this in vivo assay. When 6−10 were compared in vitro in the 3T3-L1 cell line to 2, for their ability to augment insulin-stimulated lipogenesis, 10 was again the most potent compound with 6, 7, and 9 roughly equivalent to 2. These data suggest that metabolites 6, 7, and 9 are likely to contribute to the pharmacological activity of pioglitazone (2), as had been previously reported for ciglitazone (1).
PATENT
WO 2015150476
Compound 5-[4-[2-(5-(1 -hydroxyethyl)-2-pyridinyl)ethoxy]benzyl]-2,4-thiazolidinedione of formula (1 ) can be prepared according to Scheme 1 (see e.g. J.Med.Chem. 1996, 39(26),5053).
Yet another method to prepare mixtures (c) – comprising compound (2) and (4) – and (d) – comprising compounds (3) and (5) – (scheme 3), includes the resolution of the racemic mixture VIII using the already described methods (chiral HPLC separation, enzymatic resolution, chiral resolution, etc) followed by double bond reduction in each of the enantiomers Villa and Vlllb.
Scheme 4
Compounds of formula (2), (3), (4) and (5) may be obtained from mixtures (c) and (d) (Scheme 45) by chiral HPLC separation. Alternatively, the desired enantiomerically pure compounds can be prepared by chiral synthetic procedures known to those skilled in the art (for example: asymmetric hydrogenolysis of the corresponding single isomer of compound VI).
HPLC Method
Column: Symmetry Shield RP-18, 5 μηη (4.6 x 250 mm); wavelength: 210 nm; flow: 1 mL/min; run time: 28 min; mobile phase-gradient: (t/%B): 0/10, 8/10, 12/60, 16/80, 20/80, 24/10, 28/10 [A: Water (potassium dihydrogen o-phosphate (pH~3)), B: Acetonitrile]
A mixture of compounds (2) and (4) (mixture (c)) and a mixture of compounds (3) and (5) (mixture (d)) were prepared according to Scheme 7.
Example 6: Preparation of diastereomeric mixtures D-1 and D-2 of M-IV:
Scheme 1 :
Ent-1 (VIII) Ent-2 (VIII)
Step 3 Step 3
MIV D-1 MIV D-2
Step 1 : Synthesis of compound VIII: HCI (48 ml, 2N) was added to a solution of compound VI (10 g, 0.024 mol) in methanol (200 ml) and the mixture was heated to reflux. After 4 h of reflux, the reaction mixture was cooled to r.t. and concentrated under reduced pressure to afford a yellow solid. The solid was suspended in water (70 ml) and neutralized using a saturated NaHC03 solution. The resulting pale yellow precipitate was collected by filtration and vacuum dried to afford compound VIII (7.5 g; 84% yield).
ES-MS [M+1]+: 371.0.
Step 2: Chiral prep. HPLC
Compound VIII (1 .0 g) was dissolved in a mixture containing equal volumes of acetonitrile, methanol and dichloromethane; injected (150 μΙ injections) in chiral prep-HPLC column (Chiralpak-IA 250 x 20 mm, 5 micron) and separated [Mobile phase- n-Hexane/0.05% Et3N in EtOH (50:50); flow Rate: 18ml/min; run time: 60 min]. The fractions containing the enantiomers Villa and Vlllb were separately concentrated under reduced pressure to minimum volume and the respective residues were diluted with EtOAc (100 ml), followed by water (50 ml). The resultant organic phases were
dried over anhydrous Na2S04 and concentrated to afford compounds Villa and Vlllb as off-white solids. Enantiomers Villa and Vlllb were isolated but the absolute configuration of each enantiomer has not been determined.
Step 3: A solution of NaBH4 (77 mg, 2.02 mmol) in 0.1 N NaOH (2 ml) was added slowly to a stirred solution of compound Ent-1 (VIII) (250 mg, 0.675 mmol), dimethylglyoxime (32 mg, 0.27 mmol) and CoCI2.6H20 (16 mg, 0.067 mmol) in a mixture of water (10 ml), THF (10 ml) and 1 M NaOH (0.5ml) solution at 10 °C, and the reaction mixture was stirred at r.t. for 1 h. After color of the reaction medium faded, additional quantity of NaBH4 (26 mg, 0.675 mmol) and CoCI2.6H20 (16 mg, 0.067 mmol) were added and stirring was continued at r.t. [additional quantities of CoC|2 and NaBH4 were added at 12 h intervals till the starting material was consumed, as monitored by LCMS]. After 90-96 h, the reaction mixture was neutralized with AcOH (pH~7); diluted with water (10 ml) and extracted in EtOAc (3 χ 50 ml). The combined organic extract was dried over anhydrous Na2S04 and concentrated to afford crude compound which was purified by flash column chromatography (Si02; 4% methanol in CH2CI2) to afford diastereomeric mixture of MIV D-1 (125 mg) as off-white solid.
Synthesis of D-2 MIV
Step 3: A solution of NaBH4 (72 mg, 1 .921 mmol) in 0.1 N NaOH (2 ml) was added slowly to a stirred solution of compound Ent-2 (VIII) (237 mg, 0.64 mmol), dimethylglyoxime (30 mg, 0.256 mmol) and CoCI2.6H20 (15 mg, 0.064 mmol) in a mixture of water (10 ml), THF (10 ml), and 1 M NaOH (0.5ml) solution at 10 °C, and the
reaction mixture was stirred at r.t. for 1 h. After color of the reaction medium faded, additional quantity of NaBH4 (24 mg, 0.64 mmol) and CoCI2.6H20 (15 mg, 0.064 mmol) were added and stirring was continued at r.t. [additional quantities of CoCI2.6H20 and NaBH4 were added at 12 h intervals till the starting material was consumed, as monitored by LCMS]. After 96 h, the reaction mixture was neutralized with AcOH (pH~7); diluted with water (10 ml) and extracted in EtOAc (3 χ 50 ml). The combined organic extract was dried over anhydrous Na2S04 and concentrated to afford crude compound, which was purified by flash column chromatography (Si02; 4% methanol in CH2CI2) to afford diastereomeric mixture of MIV D-2 (100 mg) as off-white solid.
Diastereomeric mixtures D-1 and D-2 of MIV correspond to mixtures (c) and (d) described above, but the specific diastereomers present in each diastereomeric mixture have not been assigned.
Example 7: in vitro ADME and toxicological characterization
Protocol: The assays performed include cytochrome P450 inhibition with the different isoforms, microsomal and hepatocyte stability, neurotoxicity in neural cells and hERG safety assays using a patch clamp electrophysiology measurement (FDA Draft Guidance for Industry. Drug Interaction Studies – Study Design, Data Analysis, Implications for Dosing, and Labelling Recommendations 2012, The European Medicines Agency (EMA) Guideline on the Investigation of Drug Interactions Adopted in 2012, Schroeder K et al. 2003 J Biomol Screen 8 (1 ); 50-64, Barter ZE et al. 2007
Curr Drug Metab 8 (1 ); 33-45, LeCluyse EL and Alexandre E 2010 Methods Mol Biol 640; 57-82). The results indicate a safe and favourable ADME profile for the compounds of the invention.
Example 8: The brain plasma ratios of Pioglitazone, MIV, Mill and Mil following oral dosing of a single administration of Pioglitazone at 4.5 mg/kg in male C57BL/6 mice.
The brain-plasma ratio was calculated based on levels of Pioglitazone, MIV, Mill and Mllin plasma and brain quantified at C max (maximal concentration) following oral dosing of a single administration of Pioglitazone at 4.5 mg/kg in male C57BL/6 mice. The percentage brain plasma ratio was 9, 13, 7 and 1 %, respectively, for Pioglitazone, Mil and Mill as shown in the Figure 4. Thus, active metabolites Mill and Mil crossed the BBB at much lower extent than Pioglitazone as it was predicted based on the physicochemical properties of the compounds (see Tablel ). In contrast, unexpectedly metabolite MIV crossed the BBB in a higher percentage than the parent compound Piolgitazone
The calculations of the both indexes (ClogP and QPIogBB) for Pioglitazone and its metabolites Mil and Mill are shown in Table 1 . For both indexes the 2 metabolites are lower than for pioglitazone, suggesting for Mil, and Mill a less favored penetration and distribution within CNS.
TABLE 1
PATENT
WO 2018116281
https://patents.google.com/patent/WO2018116281A1/enPioglitazone is a “dirty” drug which is converted to many metabolites in vivo. The metabolic pathway of pioglitazone after oral administration has been studied in several animal species and in humans and the metabolites have been described in the literature (see e.g. Sohda et al, Chem. Pharm. Bull., 1995, 43(12), 2168-2172) and Maeshiba et al, Arzneim.-Forsch/Drug Res, 1997, 47 (I), 29-35). At least six metabolites have been identified, named M-I to M-VI. Amongst these metabolites, M-II, M-III and M-IV show some pharmacological activity but are less active than Pioglitazone in diabetic preclinical models.
[0005] 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione has the following structure:
[0006] Tanis et al. (J. Med. Chem. 39(26 ):5053-5063 (1996)) describe the synthesis of 5-[[4-[2-[5-( 1 -hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione as follows:Scheme 1
[0007] Tanis et al. describe that the intermediate 14 was obtained in a 27% yield by reacting compound 13 in an aqueous 37% formaldehyde at 170°C for 6 hours. In this process, 5-[[4- [2-[5-( 1 -hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione (compound 6 in Scheme 1) was obtained in a 2.47% overall yield.[0008] WO 2015/150476 Al describes the use of 5-[[4-[2-[5-(l-hydroxyethyl)-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione, and its pharmaceutically acceptable salts, in the treatment of central nervous system (CNS) disorders. WO 2015/150476 Al describes that 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione was prepared according to the process of Tanis et al. (supra) where the intermediate corresponding to compound 14 of Tanis et al. was prepared similarly at 160°C for 5 hours providing a 17% yield. The overall yield of 5-[[4-[2-[5-(l-hydroxyethyl)-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione was about 1.5%.[0009] Due to the low yield of the intermediate 2-[5-(l-methoxymethoxy-ethyl)pyridine-2- yl]ethanol, the process step for preparing this intermediate is critical for the overall yield of the product, 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione. In addition, the prior art process to obtain compound 14 is difficult to scale because the reaction is carried out in a pressure vessel at a very high temperature and it is a very dirty reaction.[0010] Accordingly, the processes described in the art afford the product 5-[[4-[2-[5-(l- hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione only in a very low overall yield and, therefore, they are not suitable for large scale synthesis. In addition, the prior art process employs CH3OCH2CI, a known carcinogen, for protecting the hydroxyl group in the key intermediate. There is a need for an improved process for synthesizing 5- [[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione, and its pharmaceutically acceptable salts.Formula I illustrated by Scheme 2:Scheme 2 r
B
deprotectionoptional saltformation
I (HCI salt)[0255] In another embodiment, the disclosure provides a process for preparing the compound of Formula I illustrated by Scheme 3 : Scheme 3C
Br. e
step ‘< step b step c
step step g
[0256] In another embodiment in Scheme 3, step c, the order of mixing of the reagents can be as follows: 1. n-BuLi, 2. ethylene oxide, and 3. Cul. This order of mixing is described in Example 2.[0257] In the step a, 2,5-dibromopyridine (1) is reacted with i-PrMgCl in THF and then further with acetaldehyde to obtain compound 2. The reaction mixture is preferably filtered over Celite® after the reaction to remove most of the salts. In one embodiment, the addition of acetaldehyde is conducted at a temperature between -15°C and -10°C to control the exothermic reaction. [0258] In the step b, compound 2 is reacted with TBDMS-C1 in the presence of imidazole having DMF as a solvent. The crude product 3 is advantageously purified by a short plug filtration.[0259] In the step c, the hydroxyl protected compound 3 is reacted with ethylene oxide in the presence of n-BuLi and Cu(I)iodide while maintaining the reaction temperature, i.e., the reaction mixture temperature, below -20°C. In one embodiment, the reaction temperature is maintained below -55°C while adding n-BuLi and Cu(I)iodide into the reaction mixture. In another embodiment, the temperature of the reaction mixture is maintained below -55°C while adding n-BuLi, followed by ethylene oxide and then Cu(I)iodide into the reaction mixture. In another embodiment, the temperature of the reaction mixture is maintained below -55°C while adding n-BuLi into the reaction mixture, followed by ethylene oxide. In this embodiment, Cu(I)iodide is added then into the reaction mixture while the reaction mixture temperature is maintained below -20°C, and preferably below -55 °C. The reaction mixture is then allowed to slowly warm to room temperature after the addition of the reagents and stirred at room temperature, e.g., 20-25°C, overnight. This process is described in detail in Example 2. After the reaction, the complexed copper is advantageously removed by washing with 10% ammonia. The crude compound 4 can be purified by column chromatography to give >99% pure product with a yield of about 52%.[0260] The following examples are illustrative, but not limiting, of the methods of the present invention. Suitable modifications and adaptations of the variety of conditions and parameters normally encountered in clinical therapy and which are obvious to those skilled in the art in view of this disclosure are within the spirit and scope of the invention.ExamplesCOMPARATIVE EXAMPLE 1Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]- 2,4-thiazolidinedione (9a) according to the process described in WO 2015/150476 Al Scheme 4
8a 9a[0261] (a) Synthesis of l-(6-methyl-pyridin-3-yl)-ethanol (3a)[0262] LiHMDS (1.0 M in tetrahydrofuran, 463 ml, 0.463 mol) was added drop wise to a cooled solution of methyl 6-methylnicotinate (la) (20 g, 0.132 mol) and ethyl acetate (82 g, 0.927 mol) in dimethylformamide at -50°C; gradually raised the temperature to room temperature and stirred at the same temperature. After 1 h, the reaction mixture was cooled to 0°C; slowly diluted with 20% sulphuric acid and heated to reflux. After 4 h, the reaction mixture was cooled to room temperature, and further to 0°C and basified with potassium carbonate. The reaction medium was diluted with water and extracted in ethyl acetate (3×50 mL). Combined organic extract was dried over sodium sulphate and concentrated to afford crude l-(6-methylpyridin-3-yl)ethan-l-one (2a) (20.0 g) which was taken to the next step without any purification. ES-MS [M+l]+: 136.1.Sodium borohydride (2.3 g, 0.06 mol) was added in small portions over 30 min, to a solution of compound 2a (16.4 g, 0.121 mol) in ethanol (160 mL) at 0°C and the reaction mixture was stirred at same temperature. After 1 h, the reaction mixture was diluted with sodium bicarbonate solution (sat) (2×200 mL) and extracted with dichloromethane (2×500 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford a pale yellow oil, which was purified by flash column chromatography (5% methanol/dichloromethane) to afford compound 3a (17.0 g; 93% yield over 2 steps) as a pale yellow oil. ES-MS [M+l]+: 138.1. 1H NMR (400 MHz, CDC13): δ 8.35 (d, J = 2.0 Hz, 1H), 7.63 (dd, J = 8.0, 2.4 Hz, 1H), 7.12 (d, J = 8.0 Hz, 1H), 4.89 (q, J = 6.5 Hz, 1H), 3.30 (br s, 1H), 2.50 (s, 3H), 1.48 (d, J = 6.5 Hz, 3H).[0263] (b) Synthesis of 5-(l-methoxymethoxy-ethyl)-2-methyl-pyridine (4a):Compound 3a (15 g, 0.109 mol) was added, drop wise, to a cooled suspension of sodium hydride (6.56 g, 0.164 mol) in tetrahydrofurane (150 mL) and stirred at 0°C. After 30 min, chloromethyl methyl ether (13.2 g, 0.164 mol) was added drop wise while stirring and keeping the internal temperature around 0°C. After addition is over, the reaction mixture was stirred at the same temperature for 1 h. The reaction was quenched with ice cold water (80 mL) and extracted with ethyl acetate (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford an orange color oil, which was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 4a (10.0 g; 51% yield) as a pale yellow oil. ES-MS [M+l]+: 182.2. 1H NMR (400 MHz, CDC13): δ 8.45 (d, J = 2.0 Hz, 1H), 7.56 (dd, J = 8.0, 2.0 Hz, 1H), 7.14 (d, J = 8.0 Hz, 1H), 4.75 (q, J = 6.4 Hz, 1H), 4.57 (ABq, 2H), 3.36 (s, 3H), 2.53 (s, 3H), 1.48 (d, J = 6.6 Hz, 3H).[0264] (c) Synthesis of 2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethanol (5a):A mixture of compound 4a (7.0 g, 0.0386 mol) and 37% formaldehyde solution (5.8 g, 0.077 mol) was heated to 160°C in a sealed glass tube for 5 h. The reaction mixture was cooled to room temperature and concentrated under reduced pressure to afford a crude compound which was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 5 (1.2 g; 17% yield) as pale yellow oil. ES-MS [M+l]+: 212.1. 1H NMR (400 MHz, CDC13): δ 8.42 (d, J = 2.0 Hz, 1H), 7.65 (dd, J = 8.0, 2.4 Hz, 1H), 7.25 (d, J = 8.0 Hz, 1H), 4.72 (q, J = 6.6 Hz, 1H), 4.65 (t, J = 5.6 Hz, 1H), 4.52 (ABq, 2H), 3.73 (m, 2H), 3.24 (s, 3H), 2.86 (t, J = 7.2 Hz, 2H), 1.49 (d, J = 6.4 Hz, 3H).[0265] The total yield for compound 5a from compound la was 8% molar.[0266] (d) Synthesis of 4-{2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethoxy}- benzaldehyde (6a): Methanesulphonylchloride (1.19 g, 0.01 mol) was added, drop wise, to a cooled suspension of compound 5a (1.7 g, 0.008 mol) and triethylamine (1.79 ml, 0.013 mol) in dichloromefhane (20 mL) at 0°C and stirred at same temperature for 1 h. The reaction mixture was diluted with water (50 mL) and extracted with dichloromethane (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford 2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethyl methanesulfonate (2.04 g; 88% yield) as a yellow oil, which was taken to next step without purification. ES-MS [M+l]+: 290.[0267] 2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethyl methanesulfonate was added (2.3 g, 0.008 mol) to a stirred suspension of 4-hydroxybenzaldehyde (1.65 g, 0.0137 mol) and potassium carbonate (1.86 g, 0.0137 mol) in mixture of toluene (25 mL) and ethanol (25 mL); stirred at 85°C for 5 h. After consumption of the starting materials, the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate (2×100 mL). The combined organic extract was washed with water; dried over anhydrous sodium sulphate and concentrated to afford a crude dark yellow liquid. The crude was purified by flash column chromatography (1% methanol/dichloromethane) to afford compound 6a (1.5 g; 60% yield) as pale yellow liquid. ES-MS [M+l]+: 316.1.[0268] (e) Synthesis of 5-(4-{2-[5-(l-methoxymethoxy-ethyl)-pyridin-2-yl]-ethoxy}- benzylidene)-thiazolidine-2,4-dione (7a):Piperidine (80 mg, 0.95 mmol) was added to a solution of compound 6a (0.6 g, 1.9 mmol) and thiazolidine-2,4-dione (0.22 g, 1.9 mmol) in ethanol (15 mL) and the mixture was heated to reflux overnight. After 15 h, the reaction mixture was cooled to room temperature and concentrated under reduced pressure to afford crude mixture, which was purified by flash column chromatography (2% methanol/dichloromethane) to afford compound 7 (500 mg; 64% yield) as a yellow solid. ES-MS [M+l]+: 415.1. 1H NMR (400 MHz, DMSO-d6): δ 12.25 (br s, 1H), 8.47 (d, J = 2.0 Hz, 1H), 7.70 (dd, J = 8.0, 2.0 Hz, 1H), 7.54 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 8.0 Hz, 1H), 7.21 (d, J = 8.8 Hz, 2H), 4.73 (m, 1H), 4.60-4.40 (m, 4H), 4.22 (t, J = 6.2 Hz, 1H), 3.24 (s, 3H), 3.20 (t, J = 6.8 Hz, 2H), 1.41 (d, J = 6.0 Hz, 3H).[0269] (f) Synthesis of 5-(4-{2-[5-(l-hydroxy-ethyl)-pyridin-2-yl]-ethoxy}-benzyl)- thiazolidine-2,4-dione (9a): [0270] A solution of sodium borohydride (115 mg, 3.017 mmol) in 0.2N sodium hydroxide(1.2 mL) was added slowly to a stirred solution of compound 7 (0.5 g, 1.207 mmol), dimethylglyoxime (42 mg, 0.36 mmol) and C0CI2.6H2O (23 mg, 0.096 mmol) in a mixture of water (6 mL): tetrahydrofurane (6 mL) and 1M sodium hydroxide (1 mL) solution at 10°C and after addition, the reaction mixture was stirred at room temperature. After 1 h, the reaction color lightened and additional quantities of sodium borohydride (46 mg, 1.207 mmol) and C0CI2.6H2O (22 mg, 0.096 mmol) were added and stirring was continued at room temperature. After 12 h, the reaction was neutralized with acetic acid (pH~7); diluted with water (10 mL) and extracted in ethyl acetate (3×50 mL). The combined organic extract was dried over anhydrous sodium sulphate and concentrated to afford crude compound 8a, 5-(4- (2-(5-(l-(methoxymethoxy)ethyl)pyridin-2-yl)ethoxy)benzyl)thiazolidine-2,4-dione, (0.4 g) as pale yellow semi solid, which was taken to next step without purification. ES-MS [M+l]+: 417.5.[0271] 2N HC1 (2 mL) was added to a solution of compound 8a (0.4 g, 0.96 mmol) in methanol (20 ml) and the mixture was heated to reflux. After 4 h, the reaction mixture was cooled to room temperature and then concentrated under reduced pressure to afford a residue which was dissolved in water and the solution was neutralized using sodium bicarbonate solution (sat). The resulting white precipitate was collected by filtration to afford compound 9a (250 mg; 56% yield over 2 steps) as an off-white solid. ES-MS [M+l]+: 373.4. 1H NMR (400 MHz, DMSO-de): δ 12.00 (br s, -NH), 8.46 (d, J = 2.0 Hz, 1H), 7.66 (dd, J = 8.0, 2.4 Hz, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.13 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 2H), 5.25 (d, J = 4.4 Hz, 1H), 4.86 (m, 1H), 4.75 (m, 1H), 4.30 (t, J = 6.8 Hz, 2H), 3.30 (m, 1H), 3.14 (t, J = 6.4 Hz, 2H), 3.04 (m, 1H), 1.34 (d, J = 6.4 Hz, 3H).[0272] The overall yield of compound 9a was 1.5% molar.EXAMPLE 2Synthesis of 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol[0273] The synthesis of 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol was conducted according to the Scheme 5 using the reagents and solvents listed in Table 1 below: Scheme 5TBDMS-CI OTBDMS 1 . n-BuLi, <-55°C OTBDMSImidazole
DMF
[0274] The 1H-NMR spectra were recorded with Agilent MercuryPlus 300 NMR spectrometer.[0275] LC-MS data were obtained on an Agilent 1290 series with UV detector and HP 6130MSD mass detector using as column Waters XB ridge BEH XP (2.1 x 50 mm; 2.5 μιτι) and as eluent Ammonium acetate (10 mM); Water/ Methanol/ Acetonitrile.[0276] (a) l-(6-bromopyridin-3-yl)ethan-l-ol (2)[0277] A 20 L vessel was placed under nitrogen atmosphere and charged with tetrahydrofuran (5.5 L) and 2,5-dibromopyridine (1) (2000 g, 8.44 mol, 1.0 eq) (OxChem Corporation). The mixture was cooled to -10°C and isopropyl magnesium chloride (20% in THF, 6.02 L, 11.82 mol, 1.4 eq) (Rockwood Lithium) was added slowly over 1 h, keeping the reaction temperature below 5°C. After addition, the cooling bath was removed and the temperature was kept below 30°C (some additional cooling was needed to achieve this) and the reaction mixture was stirred overnight. After 16 h, a sample was taken; quenched with saturated aqueous ammonium chloride and extracted with methyl tert-buty\ ether (TBME). The TBME was evaporated under vacuum. 1H-NMR in deuterated chloroform showed complete conversion.[0278] The reaction mixture was cooled to -15°C and a solution of acetaldehyde (472 g,10.72 mol, 1.27 eq) (Acros) in tetrahydrofuran (200 mL) was added dropwise, while keeping temperature below -10°C. After the addition was complete, the cooling bath was removed and the temperature was allowed to rise to maximum of 5-8°C. After 1.5 h, a sample was taken and the reaction was quenched with aqueous ammonium chloride as described above. 1H-NMR showed the reaction was complete.[0279] Two batches were combined for work up.[0280] The reaction mixture was quenched by pouring the mixture into a solution of aqueous ammonium chloride (1 kg in 5 L water) and stirred for 15 min, filtered over Celite and rinsed thoroughly with toluene. The filtrate was transferred to a separation funnel and the obtained two layers system was separated. The aqueous layer was extracted with toluene (2 L). The combined organic layers were dried over sodium sulfate and filtered. Evaporation of the filtrate to dryness under vacuum yielded 3.49 kg (99%) of the desired crude material. XH NMR (300 MHz, CDC13): δ 8.30 (d, J = 2.5 Hz, 1H), 7.59 (dd, J = 8.0, 2.5 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 4,91 (q, J = 6.5 Hz, 1H), 1.49 (d, J = 6.5 Hz, 3H).[0281] (b) 2-bromo-5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridine (3)[0282] A 50 L reactor under nitrogen atmosphere was charged with compound 2 (10.0 kg, around 49.5 mol) and DMF (16 L). The mixture was cooled to 10°C and imidazole (6.74 kg, 99 mol, 2.0 eq) (Apollo Scientific Ltd.) was added portion wise within 30 min. The mixture was cooled to 0°C and TBDMS-Cl (7.46 kg, 49.5 mol, 1.0 eq) (Fluorochem) was added portion wise within 5 h, keeping the temperature below 3°C. The mixture reaction temperature was allowed to reach room temperature and stirred overnig ht. H NMR of a sample showed complete conversion.[0283] The reaction mixture was transferred to a 100 L extraction-vessel and the product was extracted with heptane (2×7.5 L, 10 L). The combined heptane-layers were washed with water (2×6 L, 3 L) to remove small amounts of DMF, dried over sodium sulfate and evaporated under vacuum to give crude compound 3 (15.5 kg, 49.0 mol) in a 99.0% yield. This crude product was purified by a short plug filtration, using 10 kg silica/heptane and eluted with heptane (approx. 50 L). The product-fractions were combined and evaporated under vacuum to give 12.0 kg of purified compound 3 (38 mol) as a brown oil in a 76.8% molar yield. (Average yield for 3 experiments was 78%). HPLC-MS: Rt= 2.6 min, M+l=316.1 and 318.1; 1H NMR (300 MHz, CDC13): δ 8.55 (d, J = 2.2 Hz, 1H), 7.54 (dd, J = 8.2, 2.2 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 4,86 (q, J = 6.5 Hz, 1H), 1.40 (d, J = 6.5 Hz, 3H), 0.88 (s, 9H), 0.02 (d, J = 26 Hz, 2x3H).[0284] (c) 2-(5-(l-((tert-butyldimethylsilyl)oxy)ethyl)pyridin-2-yl)ethan-l-ol (4)[0285] The ethylene oxide solution in diethylether was prepared in advance. Diethylether(1.2 L) in a 3 L three-necked flask was cooled at -65 °C and ethylene oxide (462.3 g, 10.5 mol, 1.06 eq) (Linde) was added and stirred at -70°C. Alternatively, the ethylene oxide solution can be made at about -20°C and then added gradually to the reaction mixture having a temperature at about -60°C. [0286] To a solution of 2-bromo-5-(l-((ieri-butyldimethylsilyl)oxy)ethyl)pyridine (3) (3.13 kg, 9.90 mol, 1.0 eq) in diethylether (7.5 L) cooled at -59°C, n-butyllithium (4 L, 10.0 mol, 2.5M in hexanes, 1.01 eq) (Aldrich Chemistry) was added while keeping temperature between -58°C and -62°C. After addition, the mixture was stirred for 1 h while keeping temperature between -60°C and -68°C. The upfront prepared ethylene oxide solution was added at once to the reaction mixture, while temperature was around -62°C. Subsequently, copper(I) iodide (962.3 g, 5.05 mol, 0.51 eq) (Acros Organics) was added in portions of 120 g, every 10 min, keeping the temperature between -61°C and -63°C. Stirring was continued for 1 h after addition keeping temperature between -61°C and -63°C. The cooling bath was removed and allowing the temperature to rise to about 15°C and further to 25 °C with a water bath overnight.[0287] Workup: The reaction-mixture was poured into a solution of 1 kg ammonium- chloride in 5 L water and stirred for 30 min, then the layers were separated. The organic layer was washed with aqueous ammonium hydroxide (10%, 2.5 L, 4x) to remove Cu-complex (blue color disappeared). The combined organic layers were dried over sodium sulfate and evaporated to give 3.12 kg (max. 9.90 mol) crude compound 4 as a brown oil. The crude compound was purified over 20 kg silica (heptane/EtOAc) by eluting with 80 L heptane/EtOAc, 20 L EtOAc, 25 L EtOAc/MeOH 95/5, 25 L EtOAc/MeOH 9/1 and 10 L EtOAc/MeOH 8/2, to give 1.47 kg of purified compound 4 (5.22 mol) as a brown oil (with tendency to solidify) in a 52.7% average molar yield (HPLC-purity of 99.5%). (Average yield over 12 experiments 52%). HPLC-MS: Rt= 2.3 min, M+l=282.1; 1H NMR (300 MHz, CDC13): δ 8.42 (d, J = 2.1 Hz, 1H), 7.61 (dd, J = 8.3, 2.1 Hz, 1H), 7.11 (d, J = 8.3 Hz, 1H), 4,88 (q, J = 7.0 Hz, 1H), 4.01 (t, J=6.0 Hz, 2 H), 3.00 (t, J=6.0 Hz, 2 H), 1.41 (d, J =7.0 Hz, 3H), 0.90 (s, 9H), 0.02 (d, J = 26 Hz, 2x3H).[0288] Another 2.5% of the product was isolated by re -purifying impure product fraction.The total yield of compound 4 from compound 1 was 39.6% molar.EXAMPLE 3Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]- 2,4-thiazolidinedione hydrochloride (9) 2. Sodium bisulfiteethanol/water mixture
3. Addition 10% aqueous sodium hydroxide solution
until pH 12
from step e dimethylglyoxime7step g step f
step h[0289] The 1H-NMR spectra were recorded with a 400 MHz Avance Bruker NMR spectrometer. LC-MS data were obtained on a Agilent Technologies 6130 Quadrapole LC/MS using as column Agilent XDB-C18 and as eluent 0.1% formic acid (aq) and 0.05% formic acid in acetonitrile.[0290] Steps d and e: Synthesis of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl] ethoxy] -benzaldehyde (6)[0291] To a well stirred solution of 5-[[[(l,l-dimethylethyl)dimethylsilyl]-oxy]ethyl]-2- pyridineethanol (4) (obtained as described in Example 2) (1.91 kg) in toluene (8.6 L) at 5°C were added sodium hydroxide (30% aqueous, 2.79 L) and tetrabutylammonium bromide (7.2 g). p-Toluenesulfonyl chloride (1.62 kg) was next added in portions during 5 min. After the addition, the reaction mixture was allowed to reach room temperature in 0.5 h and stirred at this temperature for 18 h. Water (7.3 L) was then added and the mixture was mixed well. Once the solids were dissolved, the layers were allowed to settle and the organic layer was separated. This organic phase was washed with water (5.7 L, 2x), followed by washing with a solution of sodium chloride (57 g) in water (5.7 L). The solvents were concentrated at reduced pressure to an amount of 2.5 kg of a brown oil (compound 5).[0292] To this well stirred brown oil were added subsequently ethanol (7.8 L), water (0.86L), 4-hydroxybenzaldehyde (0.88 kg) and potassium carbonate (1.17 kg) and then the mixture was heated at 75 °C for 18 h. Then, the solvent was evaporated while adding toluene (7.7 L) during 6 h and then the reaction mixture was allowed to cool. At 30°C, water (7.6 L) was added, stirred until all solids were dissolved and the mixture was cooled to room temperature. The layers were allowed to settle and separated. The organic layer was washed with water (7.6 L). The first aqueous extract was extracted with toluene (2.8 L) and this organic extract was used to also extract the aqueous washing. The organic extracts were combined and concentrated under vacuum to give 3.49 kg of a black oil (crude title compound 6).[0293] 1.73 kg of this black oil was dissolved in ethanol (0.74 L) and added to a well stirred solution of sodium bisulfite (1.36 kg) in a mixture of water (3.27 L) and ethanol (0.74 L). The container of the black oil was rinsed with ethanol (0.37 L) twice and these two rinses were also added to the bisulfite reaction mixture. After 75 min, heptane (5.3 L) was added, well mixed for 5 min, and the layers were allowed to settle and separated. To the organic layer was added a solution of sodium bisulfite (0.55 kg) in water (2.65 L), and ethanol (1.06 L). After stirring for 30 min, the layers were allowed to settle and separated. The two bisulfide aqueous extracts were combined and flasks rinsed with water (2.12 L). Next, toluene (4.5 L) and heptane (4.5 L) were added, the mixture was well stirred and the pH was adjusted to 12 using sodium hydroxide (10% aq) (temperature became 32°C). After stirring for an additional 5 min, the layers were allowed to settle and separated at 30°C. The aqueous layer was extracted with a mixture of toluene (1.5 L) and heptane (3.0 L). The layers were separated and the organic layers were combined. The combined organic layers were washed with water (5 L, 2x) and concentrated under vacuum to give the purified title compound 6. This procedure was repeated with another 1.73 kg of the black oil (crude title compound 6) to give in total 2.77 kg of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl]ethoxy]-benzaldehyde (6) as brown oil which contained 24% m/m of toluene according to 1H NMR (yield = 80%, calculated from compound 4 and corrected for residual toluene). [0294] 1H NMR (CDC13) δ: 0.00 (s, 3H), 0.09 (s, 3H), 0.91 (s, 9H), 1.44 (d, = 6 Hz, 3H),3.30 (t, = 7 Hz, 2H), 4.47 (t, = 7 Hz, 2H), 4.92 (q, = 6 Hz, 1H), 6.99 – 7.30 (m, 3H), 7.62- 7.67 (m, 1H), 7.80 – 7.85 (m, 2H), 8.5- 8.54 (m, 1H) and 9.88 (s, 1H).[0295] LC-MS; rt 7.5 min: ES: M+ 387, 386.[0296] Step f: Synthesis of (5Z)-5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methylene]-2,4-thiazolidinedione (7)[0297] A solution of 4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]- ethoxy]-benzaldehyde (6) (2.75 kg, containing 24% m/m of toluene) and piperidine (6.0 g) in methanol (3.16 L) was concentrated at 40°C under reduced pressure. The residue was dissolved in methanol (10.4 L) and 2,4-thiazolidinedione (759 g) and piperidine (230 g) were added. The mixture was heated at 47°C. After 25 h, the reaction mixture was allowed to cool to room temperature. The mixture was kept at pH 5-6 by adjusting it with acetic acid, if necessary. After a night at room temperature, water (1.56 L) was added and the suspension was stirred at room temperature for additional 2 h. The solids were isolated by filtration, washed with methanol (1 L, 2x) and dried under vacuum to give crude compound 7 (1.65 kg). The crude compound was mixed with methanol (10 L) and dichloromethane (8.6 L) and heated at 32°C until all solids dissolved. Then, the solvents were removed by distillation until the temperature of the mixture reached 34°C at a pressure of 333 mbar. Then, it was allowed to cool to room temperature overnight and stirred at 2°C for additional 2 h. The solids were isolated by filtration, washed with methanol (0.5 L, 2x) and dried under vacuum to give title compound 7 (1.50 kg) (yield = 61%).[0298] 1H NMR (CDCI3) δ 0.00 (s, 3H), 0.08 (s, 3H), 0.90 (s, 9H), 1.43 (d, = 6 Hz, 3H),3.32 (t, = 7 Hz, 2H), 4.48 (t, = 7 Hz, 2H), 4.92 (q, = 6 Hz, 1H), 6.95 – 7.00 (m, 2H), 7.24 – 7,28 (m, 1H), 7.38 – 7.42 (m, 2H), 7.67 (s, 1H), 7.69 – 7.73 (m, 1H) and 8.48 (d, = 3 Hz, 1H).[0299] LC-MS; rt 7.5 min: ES: M+ 487, 486, 485.[0300] Step g: Synthesis of 5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]ethyl]-2- pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione (8)[0301] To a stirred suspension of (5Z)-5-[[4-[2-[5-[[[(l,l-dimethylethyl)dimethylsilyl]oxy]- ethyl]-2-pyridinyl]ethoxy]phenyl]methylene]-2,4-thiazolidinedione (7) (10 g) in THF (10 mL) and sodium hydroxide (IN aq, 21 mL) was added of a solution of cobalt chloride (26 mg) and of dimethylglyoxime (930 mg) in THF (2.3 mL) and water (1.0 mL). Then the suspension was put under a nitrogen atmosphere by applying the sequence of vacuum and flushing with nitrogen (4x). Thereafter, the suspension was heated to 30°C. Then, a stock solution of sodium borohydride was prepared by dissolving sodium borohydride (2.7 g) in a mixture of water (15.8 mL) and a solution of sodium hydroxide (1 N aq, 3.5 mL), which was put under a nitrogen atmosphere by applying a sequence of vacuum and flushing with nitrogen (3x). This was added to the suspension of compound 7 at a rate of 4.5 mL/h. Simultaneously, nitrogen gas-saturated acetic acid was added to the suspension at a rate of 0.7 mL/h to maintain a pH of 10.0-10.5. After 1 h 30 min the rate of addition of the sodium borohydride solution and acetic acid were both reduced by half. Next, 3 h 45 min after start of addition, the addition of sodium borohydride and acetic acid were stopped. The mixture was allowed to cool down to room temperature and acetone (2.5 mL) was added over a period of 1 minute. After stirring the reaction mixture for 15 min acetic acid was added until the pH was 5.5-6.0 (about 3 mL required). Next, a mixture of ethyl acetate/toluene (1/3 v/v, 30 mL) was added, well mixed and layers were allowed to settle. The aqueous layer was separated and washed with ethyl acetate/toluene (1/3 v/v, 10 mL). Both organic extracts were pooled and water (40 mL) was added, well mixed and layers were allowed to settle. The pH of the aqueous layer was adjusted to 5.5-6 using saturated sodium hydrogen carbonate solution (aq) and again mixed with the organic layer. Layers were allowed to settle and the organic layer was separated and concentrated under vacuum to give 11.09 g of yellow oil (crude mixture containing title compound 8 and its borane complex). Several batches were combined for work up.33.1 g of the crude mixture containing title compound 8 and its borane complex (not corrected for residual solvents) was dissolved in toluene (30 mL) and filtered. The filtrate was submitted to column chromatography (silica gel, gradient of toluene to toluene/ethyl acetate 1/1) to give 30.0 g of mixture of 5-[[4-[2-[5-[[[(l,l- dimethylethyl)dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione (8) and its borane complex as a slightly yellow oil (yield = 100% from compound 4, not corrected for residual solvents). [0303] 1H NMR (CDC13) δ: -0.03 – 0.10 (m, 6H), 0.87 – 0.93 (m, 9H), 1.42 (d, / = 6 Hz, 3H),3.05-3.71 (m, 4H), 4.30 – 4.51 (m, 3H), 4.87 – 4.94 (m, 1H), 6.82 – 6.88 (m, 2H), 7.10-7.92 (m, 5H), 8.49 (d, / = 3 Hz, 0.6H) and 8.72 (brs, 0.4H).[0304] LC-MS; rt 6.8 min: ES: M+ 489, 488, 487, M“ 487, 486, 485; rt 8.1 min: ES M“ 501,500, 499, 498, 485.[0305] Step h: Synthesis of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]- methyl]-2,4-thiazolidinedione hydrochloride (9)[0306] To a stirred solution of the mixture of (5-[[4-[2-[5-[[[(l,l-dimethylethyl)- dimethylsilyl]oxy]ethyl]-2-pyridinyl]ethoxy]phenyl]methyl]-2,4-thiazolidinedione and its borane complex (8) (5.17 g) in methanol (25.2 mL) at 22°C was added hydrochloric acid (30%, 2.75 mL) in about 5 min to give a temperature rise to 28°C. This solution was heated to 40 °C. Three hours after addition, the 11 g of volatiles were removed under reduced pressure. Then, acetonitrile (40.3 mL) was added and the mixture was heated at reflux for 0.5 h. Next, the suspension was allowed to cool down to room temperature and stirred for 1 h at room temperature. Solids were isolated by filtration, washed with a mixture of acetonitrile/water (20/1 v/v, 10 mL) and with acetonitrile (10 mL) and dried under vacuum at 40 °C to give 4.00 g of white solids (crude 9) (yield = 77%, not corrected for residual solvents).[0307] Purification of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]methyl]-2,4- thiazolidinedione hydrochloride (9):[0308] The crude mixture of 5-[[4-[2-[5-(l-hydroxyethyl)-2-pyridinyl]ethoxy]phenyl]- methyl]-2,4-thiazolidinedione hydrochloride (3.95 g, crude 9) was dissolved in methanol/water (7/2 v/v, 80 mL) by heating it to 49°C. To this solution was added washed norit (obtained by heating a suspension of norit (6 g) in methanol/water (7/2 v/v, 90 mL) at 45°C for 1 h, then isolating the norit by filtration and washing it twice with methanol/water (7/2 v/v, 30 mL) and drying it under vacuum at 40°C). Equipment was rinsed with methanol/water (7/2 v/v, 18 mL). After 0.5 h of stirring at 46°C, the warm suspension was filtered to remove the norit and filter was washed twice with methanol/water (7/2 v/v, 18 mL). The filtrate was concentrated under vacuum at a bath temperature of 60°C to a mass of 11.8 g (1 v of compound and 2 v of water). To the suspension was added butanone (19.7 mL, 5 v) and the mixture was heated at a bath temperature of 95°C. Under distillation at a constant volume, butanone (95 mL) was added. Next, heating was stopped and the suspension was allowed to reach room temperature in about 0.5 h. Subsequently it was stirred for 0.75 h at room temperature. The solids were isolated by filtration, washed with a mixture of butanone/water (95/5 v/v, 18 mL) and butanone (18 mL) and dried under vacuum at 40°C to give 3.57 g of compound 9 as white solids (yield = 91%).[0309] 1H NMR (DMSO-de): δ 12.00 (br s, -NH), 8.71 (d, = 2.0 Hz, 1H), 8.45 (dd, = 8.3,1.7 Hz, 1H), 7.98 (d, = 8.3 Hz, 1H), 7.15 (d, = 8.7 Hz, 2H), 6.88 (d, = 8.7 Hz, 2H), 5.57 (s, OH), 4.95 (q, = 6.5 Hz, 1H), 4.86 (dd, = 8.9, 4.4 Hz, 1H), 4.40 (t, = 6.3 Hz, 2H), 3.49 (t, = 6.2 Hz, 2H), 3.29 (dd, = 14.2, 4.4 Hz, 1H), 3.06 (dd, = 14.2, 9.0 Hz, 1H), 1.41 (d, = 6.5 Hz, 3H).[0310] LC-MS; rt 3.5 min: ES: M+ 374, 373, M“ 372, 371.EXAMPLE 4Conditions tested in the preparation of compound 5 in the Step d[0311] The conditions described in Table 2 below were tested in the step d in the preparation of compound 5 from compound 4 providing a good yield of compound 5:Table 2Entry Reaction Conditions Amount of p-Ts-Cl / Eq1 Toluene/water/Bu4NBr/NaOH 1.052 1.083 1.074 1.07+0.035 1.076 Et3N / DCM 1.187 1.408 Pyridine / DCM 1.40 EXAMPLE 5Conditions tested in the preparation of compound 6 in the Step e[0312] The conditions described in Table 3 below were tested in the step e in the preparation of compound 6 from compound 5 providing a good yield of compound 6:Table 3
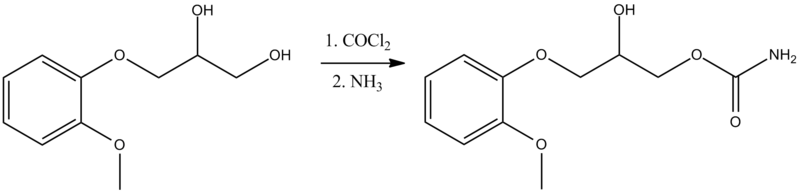
Compound 1 is administered to the subject. The structure of 5-[[4-[2-[5-(l -hydroxy ethyljpyri din-2 – yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione is:
[0047] The present disclosure encompasses the use of stereoisomers of 5-[[4-[2-[5-(l- hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione. 5-[[4-[2-[5- (l-hydroxyethyl)pyridin-2-yl]ethoxy]phenyl]methyl]-l,3-thiazolidine-2,4-dione has two asymmetric centers and thus four stereoisomers are possible as follows:
(1E)-N-{4-[(2-Hydroxybenzoyl)oxy]phenyl}ethanimidic acid118-57-0[RN] 204-261-3[EINECS] CAS Registry Number: 118-57-0 CAS Name: 2-Hydroxybenzoic acid 4-(acetylamino)phenyl ester Additional Names:p-acetamidophenyl salicylate; acetylaminophenyl salicylate; acetyl-p-aminosalol; p-acetylaminophenol salicylic acid ester; phenetsal Trademarks: Salophen (Bayer); Phenosal Molecular Formula: C15H13NO4 Molecular Weight: 271.27 Percent Composition: C 66.41%, H 4.83%, N 5.16%, O 23.59% Literature References: Prepn: Brewster, J. Am. Chem. Soc.40, 1136 (1918). Properties: Crystals from hot ethanol, mp 187°. Practically insol in petr ether, cold water, more sol in warm water. Sol in alcohol, ether, benzene. Incompatible with alkalies and alkaline solns which dissolve it with decompn. The alkaline soln gradually becomes blue when boiled, the blue color being discharged upon continued boiling and again produced upon cooling and exposure to air. Melting point: mp 187° Therap-Cat: Analgesic; antipyretic; anti-inflammatory. Therap-Cat-Vet: Analgesic; antipyretic. Keywords: Analgesic (Non-Narcotic); Anti-inflammatory (Nonsteroidal); Salicylic Acid Derivatives; Antipyretic.
It is an esterification product of salicylic acid and paracetamol. It was marketed by Bayer under the brand name Salophen as an analgesic in the late 19th and early 20th centuries.
Action and uses
In a warm alkaline solution acetaminosalol is broken up into salicylic acid and paracetamol. It is decomposed in the intestines, even when given as an injection. It was used as a substitute for salicylic acid in acute rheumatism, and as an intestinal antiseptic. It was similarly effective and much safer than salol, another intestinal antiseptic commonly used at the time. The fact that it is tasteless renders it easy to administer.Advertisement for early 20th century Bayer products, including Salophen SYNJournal of Organic Chemistry, 86(5), 4254-4261; 2021
5-[3-(1,2-Dithiolanyl)]pentanoic Acid 5-19-07-00237[Beilstein] 62-46-4[RN](+)-Thioctic acid, (+)-α-Lipoic acid, (3R)-1,2-Dithiolane-3-pentanoic acid (R)-(+)-1,2-Dithiolane-3-pentanoic acid, (R)-(+)-lipoic acid, (R)-(+)-α-Lipoic acid (R)-6,8-Dithiooctanoic acid, (R)-6,8-thioctic acid, (R)-α-Lipoic Acid, (R)-α-Lipoic Acid 1,2-Dithiolane-3-pentanoic acid, (3R)- 5-[(3R)-1,2-Dithiolan-3-yl]pentanoic acidd-Thioctic acid, (R)-(+)-alpha-Lipoic acid, (R)-(+)-Thioctic acid, Dexlipotam Thioctic Acid CAS Registry Number: 62-46-4 CAS Name: 1,2-Dithiolane-3-pentanoic acid Additional Names: 1,2-dithiolane-3-valeric acid; 6,8-thioctic acid; a-lipoic acid; 5-(1,2-dithiolan-3-yl)valeric acid; 5-[3-(1,2-dithiolanyl)]pentanoic acid; d-[3-(1,2-dithiacyclopentyl)]pentanoic acid; protogen A; acetate replacing factor; pyruvate oxidation factor Trademarks: Biletan (Gador); Thioctacid (Viatris); Thioctan (Katwijk); Tioctan (Fujisawa) Molecular Formula: C8H14O2S2, Molecular Weight: 206.33 Percent Composition: C 46.57%, H 6.84%, O 15.51%, S 31.08% Literature References: Growth factor for many bacteria and protozoa; prosthetic group, coenzyme, or substrate in plants, microorganisms, and animal tissues. Isoln of naturally occurring d-form: L. J. Reed et al.,Science114, 93 (1951); eidem,J. Am. Chem. Soc.75, 1267 (1953); Patterson et al.,ibid.76, 1823 (1954). Syntheses of dl-form: Bullock et al.,ibid.74, 1868, 3455 (1952); Hornberger et al.,ibid. 2382; Reed, US2980716 and US3049549 (1961, 1962 to Res. Corp.); Lewis, Raphael, J. Chem. Soc.1962, 4263; Ose et al.,US3223712 (1965 to Yamanouchi); J. Tsuji et al.,J. Org. Chem.43, 3606 (1978). Biosynthesis via linoleic acid: J. P. Carreau Methods Enzymol.62, 152-158 (1974). Enantioselective synthesis of d-form: P. C. Bulmanpage et al.,Chem. Commun.1986, 1408. Clinical study in treatment of Wilson’s disease: S. F. Gomes da Costa, Arzneim.-Forsch.20, 1210 (1970). Use in treatment of mushroom poisoning: R. Plotzker et al.,Am. J. Med. Sci.283, 79 (1982); J. P. Hanrahan, M. A. Gordon, J. Am. Med. Assoc.251, 1057 (1984). Reviews: Wagner, Folkers, Vitamins and Coenzymes (Interscience, New York, 1964) pp 244-263; Schmidt et al.,Angew. Chem. Int. Ed.4, 846 (1965); Schmidt et al.,Adv. Enzymol. Relat. Areas Mol. Biol.32, 423 (1969). Derivative Type: Sodium salt CAS Registry Number: 2319-84-8 Molecular Formula: C8H13NaO2S2, Molecular Weight: 228.31 Percent Composition: C 42.09%, H 5.74%, Na 10.07%, O 14.02%, S 28.09% Properties: White powder, sol in water. pH of aq solns about 7.4. Derivative Type:d-Form CAS Registry Number: 1200-22-2 Properties: Crystals by vacuum sublimation (at 85-90° and 25 microns). mp 46-48° (microblock). [a]D23 +104° (c = 0.88 in benzene). uv max (methanol): 333 nm (e 150). pKa 5.4. Practically insol in water. Sol in fat solvents.Melting point: mp 46-48° (microblock) pKa: pKa 5.4 Optical Rotation: [a]D23 +104° (c = 0.88 in benzene) Absorption maximum: uv max (methanol): 333 nm (e 150) Derivative Type:dl-Form CAS Registry Number: 1077-28-7 Properties: Yellow needles from cyclohexane, mp 60-61°. bp 160-165°. uv spectrum: Calvin, Fed. Proc.13, 703 (1954). Practically insol in water. Sol in fat solvents. Forms a water-soluble sodium salt. Melting point: mp 60-61° Boiling point: bp 160-165° Derivative Type:l-Form CAS Registry Number: 1077-27-6 Properties: Crystals from cyclohexane, mp 45-47.5° (microblock). [a]D23 -113° (c = 1.88 in benzene). uv max (methanol): 330 nm (e 140). Melting point: mp 45-47.5° (microblock) Optical Rotation: [a]D23 -113° (c = 1.88 in benzene) Absorption maximum: uv max (methanol): 330 nm (e 140) Derivative Type: Ethylenediamine Trademarks: Tioctidasi (ISI) Therap-Cat: Treatment of liver disease; antidote to poisonous mushrooms (Amanita species). Keywords: Hepatoprotectant.
Lipoic acid (LA), also known as α-lipoic acid, alpha-lipoic acid (ALA) and thioctic acid, is an organosulfur compound derived from caprylic acid (octanoic acid).[3] ALA is made in animals normally, and is essential for aerobic metabolism. It is also manufactured and is available as a dietary supplement in some countries where it is marketed as an antioxidant, and is available as a pharmaceutical drug in other countries.[3]
Physical and chemical properties
Lipoic acid (LA), also known as α-lipoic acid,[3][4] alpha-lipoic acid (ALA), and thioctic acid[5] is an organosulfur compound derived from octanoic acid.[3] LA contains two sulfur atoms (at C6 and C8) connected by a disulfide bond and is thus considered to be oxidized although either sulfur atom can exist in higher oxidation states.[3]
The carbon atom at C6 is chiral and the molecule exists as two enantiomers (R)-(+)-lipoic acid (RLA) and (S)-(-)-lipoic acid (SLA) and as a racemic mixture (R/S)-lipoic acid (R/S-LA).
LA appears physically as a yellow solid and structurally contains a terminal carboxylic acid and a terminal dithiolane ring.
“Lipoate” is the conjugate base of lipoic acid, and the most prevalent form of LA under physiological conditions.[3] Most endogenously produced RLA are not “free” because octanoic acid, the precursor to RLA, is bound to the enzyme complexes prior to enzymatic insertion of the sulfur atoms. As a cofactor, RLA is covalently attached by an amide bond to a terminal lysine residue of the enzyme’s lipoyl domains. One of the most studied roles of RLA is as a cofactor of the pyruvate dehydrogenase complex (PDC or PDHC), though it is a cofactor in other enzymatic systems as well (described below).[3]
Only the (R)-(+)-enantiomer (RLA) exists in nature and is essential for aerobic metabolism because RLA is an essential cofactor of many enzyme complexes.[3]
Biosynthesis and attachment
The precursor to lipoic acid, octanoic acid, is made via fatty acid biosynthesis in the form of octanoyl-acyl carrier protein.[3] In eukaryotes, a second fatty acid biosynthetic pathway in mitochondria is used for this purpose.[3] The octanoate is transferred as a thioester of acyl carrier protein from fatty acid biosynthesis to an amide of the lipoyl domain protein by an enzyme called an octanoyltransferase.[3] Two hydrogens of octanoate are replaced with sulfur groups via a radical SAM mechanism, by lipoyl synthase.[3] As a result, lipoic acid is synthesized attached to proteins and no free lipoic acid is produced. Lipoic acid can be removed whenever proteins are degraded and by action of the enzyme lipoamidase.[8] Free lipoate can be used by some organisms as an enzyme called lipoate protein ligase that attaches it covalently to the correct protein. The ligase activity of this enzyme requires ATP.[9]
Cellular transport
Along with sodium and the vitamins biotin (B7) and pantothenic acid (B5), lipoic acid enters cells through the SMVT (sodium-dependent multivitamin transporter). Each of the compounds transported by the SMVT is competitive with the others. For example research has shown that increasing intake of lipoic acid[10] or pantothenic acid[11] reduces the uptake of biotin and/or the activities of biotin-dependent enzymes.
The most-studied of these is the pyruvate dehydrogenase complex.[3] These complexes have three central subunits: E1-3, which are the decarboxylase, lipoyl transferase, and dihydrolipoamide dehydrogenase, respectively. These complexes have a central E2 core and the other subunits surround this core to form the complex. In the gap between these two subunits, the lipoyl domain ferries intermediates between the active sites.[3] The lipoyl domain itself is attached by a flexible linker to the E2 core and the number of lipoyl domains varies from one to three for a given organism. The number of domains has been experimentally varied and seems to have little effect on growth until over nine are added, although more than three decreased activity of the complex.[12]
The glycine cleavage system differs from the other complexes, and has a different nomenclature.[3] In this system, the H protein is a free lipoyl domain with additional helices, the L protein is a dihydrolipoamide dehydrogenase, the P protein is the decarboxylase, and the T protein transfers the methylamine from lipoate to tetrahydrofolate (THF) yielding methylene-THF and ammonia. Methylene-THF is then used by serine hydroxymethyltransferase to synthesize serine from glycine. This system is part of plant photorespiration.[13]
Biological sources and degradation
Lipoic acid is present in many foods in which it is bound to lysine in proteins,[3] but slightly more so in kidney, heart, liver, spinach, broccoli, and yeast extract.[14] Naturally occurring lipoic acid is always covalently bound and not readily available from dietary sources.[3] In addition, the amount of lipoic acid present in dietary sources is low. For instance, the purification of lipoic acid to determine its structure used an estimated 10 tons of liver residue, which yielded 30 mg of lipoic acid.[15] As a result, all lipoic acid available as a supplement is chemically synthesized.
Baseline levels (prior to supplementation) of RLA and R-DHLA have not been detected in human plasma.[16] RLA has been detected at 12.3−43.1 ng/mL following acid hydrolysis, which releases protein-bound lipoic acid. Enzymatic hydrolysis of protein bound lipoic acid released 1.4−11.6 ng/mL and <1-38.2 ng/mL using subtilisin and alcalase, respectively.[17][18][19]
Digestive proteolytic enzymes cleave the R-lipoyllysine residue from the mitochondrial enzyme complexes derived from food but are unable to cleave the lipoic acid-L–lysine amide bond.[20] Both synthetic lipoamide and (R)-lipoyl-L-lysine are rapidly cleaved by serum lipoamidases, which release free (R)-lipoic acid and either L-lysine or ammonia.[3] Little is known about the degradation and utilization of aliphatic sulfides such as lipoic acid, except for cysteine.[3]
Lipoic acid is metabolized in a variety of ways when given as a dietary supplement in mammals.[3][21] Degradation to tetranorlipoic acid, oxidation of one or both of the sulfur atoms to the sulfoxide, and S-methylation of the sulfide were observed. Conjugation of unmodified lipoic acid to glycine was detected especially in mice.[21] Degradation of lipoic acid is similar in humans, although it is not clear if the sulfur atoms become significantly oxidized.[3][22] Apparently mammals are not capable of utilizing lipoic acid as a sulfur source.
Chemical synthesis
(R)-Lipoic acid (RLA, top) and (S)-lipoic acid (SLA, down). A 1:1 mixture (racemate) of (R)- and (S)-lipoic acid is called (RS)-lipoic acid or (±)-lipoic acid (R/S-LA).
SLA did not exist prior to chemical synthesis in 1952.[23][24] SLA is produced in equal amounts with RLA during achiral manufacturing processes. The racemic form was more widely used clinically in Europe and Japan in the 1950s to 1960s despite the early recognition that the various forms of LA are not bioequivalent.[25] The first synthetic procedures appeared for RLA and SLA in the mid-1950s.[26][27][28][29] Advances in chiral chemistry led to more efficient technologies for manufacturing the single enantiomers by both classical resolution and asymmetric synthesis and the demand for RLA also grew at this time. In the 21st century, R/S-LA, RLA and SLA with high chemical and/or optical purities are available in industrial quantities. At the current time, most of the world supply of R/S-LA and RLA is manufactured in China and smaller amounts in Italy, Germany, and Japan. RLA is produced by modifications of a process first described by Georg Lang in a Ph.D. thesis and later patented by DeGussa.[30][31] Although RLA is favored nutritionally due to its “vitamin-like” role in metabolism, both RLA and R/S-LA are widely available as dietary supplements. Both stereospecific and non-stereospecific reactions are known to occur in vivo and contribute to the mechanisms of action, but evidence to date indicates RLA may be the eutomer (the nutritionally and therapeutically preferred form).[32][33]
Pharmacology
Pharmacokinetics
A 2007 human pharmacokinetic study of sodium RLA demonstrated the maximum concentration in plasma and bioavailability are significantly greater than the free acid form, and rivals plasma levels achieved by intravenous administration of the free acid form.[34] Additionally, high plasma levels comparable to those in animal models where Nrf2 was activated were achieved.[34]
The various forms of LA are not bioequivalent.[25][non-primary source needed] Very few studies compare individual enantiomers with racemic lipoic acid. It is unclear if twice as much racemic lipoic acid can replace RLA.[34]
The toxic dose of LA in cats is much lower than that in humans or dogs and produces hepatocellular toxicity.[35]
Pharmacodynamics
The mechanism and action of lipoic acid when supplied externally to an organism is controversial. Lipoic acid in a cell seems primarily to induce the oxidative stress response rather than directly scavenge free radicals. This effect is specific for RLA.[4] Despite the strongly reducing milieu, LA has been detected intracellularly in both oxidized and reduced forms.[36] LA is able to scavenge reactive oxygen and reactive nitrogen species in a biochemical assay due to long incubation times, but there is little evidence this occurs within a cell or that radical scavenging contributes to the primary mechanisms of action of LA.[4][37] The relatively good scavenging activity of LA toward hypochlorous acid (a bactericidal produced by neutrophils that may produce inflammation and tissue damage) is due to the strained conformation of the 5-membered dithiolane ring, which is lost upon reduction to DHLA. In cells, LA is reduced to dihydrolipoic acid, which is generally regarded as the more bioactive form of LA and the form responsible for most of the antioxidant effects and for lowering the redox activities of unbound iron and copper.[38] This theory has been challenged due to the high level of reactivity of the two free sulfhydryls, low intracellular concentrations of DHLA as well as the rapid methylation of one or both sulfhydryls, rapid side-chain oxidation to shorter metabolites and rapid efflux from the cell. Although both DHLA and LA have been found inside cells after administration, most intracellular DHLA probably exists as mixed disulfides with various cysteine residues from cytosolic and mitochondrial proteins.[32] Recent findings suggest therapeutic and anti-aging effects are due to modulation of signal transduction and gene transcription, which improve the antioxidant status of the cell. However, this likely occurs via pro-oxidant mechanisms, not by radical scavenging or reducing effects.[4][37][39]
All the disulfide forms of LA (R/S-LA, RLA and SLA) can be reduced to DHLA although both tissue specific and stereoselective (preference for one enantiomer over the other) reductions have been reported in model systems. At least two cytosolic enzymes, glutathione reductase (GR) and thioredoxin reductase (Trx1), and two mitochondrial enzymes, lipoamide dehydrogenase and thioredoxin reductase (Trx2), reduce LA. SLA is stereoselectively reduced by cytosolic GR whereas Trx1, Trx2 and lipoamide dehydrogenase stereoselectively reduce RLA. (R)-(+)-lipoic acid is enzymatically or chemically reduced to (R)-(-)-dihydrolipoic acid whereas (S)-(-)-lipoic acid is reduced to (S)-(+)-dihydrolipoic acid.[40][41][42][43][44][45][46] Dihydrolipoic acid (DHLA) can also form intracellularly and extracellularly via non-enzymatic, thiol-disulfide exchange reactions.[47]
RLA may function in vivo like a B-vitamin and at higher doses like plant-derived nutrients, such as curcumin, sulforaphane, resveratrol, and other nutritional substances that induce phase II detoxification enzymes, thus acting as cytoprotective agents.[39][48] This stress response indirectly improves the antioxidant capacity of the cell.[4]
The (S)-enantiomer of LA was shown to be toxic when administered to thiamine-deficient rats.[49][50]
Several studies have demonstrated that SLA either has lower activity than RLA or interferes with the specific effects of RLA by competitive inhibition.[51][52][53][54][55]
R/S-LA and RLA are widely available as over-the-counter nutritional supplements in the United States in the form of capsules, tablets, and aqueous liquids, and have been marketed as antioxidants.[3]
Although the body can synthesize LA, it can also be absorbed from the diet. Dietary supplementation in doses from 200–600 mg is likely to provide up to 1000 times the amount available from a regular diet. Gastrointestinal absorption is variable and decreases with the use of food. It is therefore recommended that dietary LA be taken 30–60 minutes before or at least 120 minutes after a meal. Maximum blood levels of LA are achieved 30–60 minutes after dietary supplementation, and it is thought to be largely metabolized in the liver.[56]
In Germany, LA is approved as a drug for the treatment of diabetic neuropathy since 1966 and is available as a non-prescription pharmaceutical.[57]
Clinical research
According to the American Cancer Society as of 2013, “there is no reliable scientific evidence at this time that lipoic acid prevents the development or spread of cancer”.[58] As of 2015, intravenously administered ALA is unapproved anywhere in the world except Germany for diabetic neuropathy, but has been proven reasonably safe and effective in four clinical trials; however another large trial over four years found no difference from placebo.[59] As of 2012, there was no good evidence alpha lipoic acid helps people with mitochondrial disorders.[60] A 2018 review recommended ALA as an anti-obesity supplement with low dosage (< 600 mg/day) for a short period of time (<10 weeks); however, it is too expensive to be practical as a complementary therapy for obesity.[61]
SYN
WO 0210151
DE 19709069; EP 0863125; US 6013833
A synthetic route based on the asymmetric reduction of oxo diesters has been reported. Meldrum’s acid (LII) was acylated by methyl adipoyl chloride (LI) in the presence of pyridine to produce the intermediate (LIII) which, upon alcoholysis with isobutanol, led to oxo diester (LIV). Enantioselective reduction of (LIV) by means of baker’s yeast furnished the (S)-hydroxy diester (LV). Alternatively, the analogous oxo diester (LVI) was prepared by acylation of methyl acetoacetate with methyl adipoyl chloride (LI), followed by deacetylation in the presence of ammonium hydroxide. Then, asymmetric chemical reduction of (LVI) by hydrogenation in the presence of the chiral catalyst Ru2Cl4[(S)-BINAP]2 provided the (S)-hydroxy diester (LVII). Regioselective reduction of either diester (LV) or (LVII) by means of NaBH4 in refluxing THF furnished dihydroxy ester (XLVIII). After conversion of (XLVIII) to the dimesylate (XLIX), displacement with potassium thioacetate afforded the bis(acetylthio) derivative (LVIII), which was further hydrolyzed with KOH to provide dihydrolipoic acid (LIX). In a related procedure, dihydrolipoic acid (LIX) was prepared by reaction of dimesylate (XLIX) with sodium disulfide, followed by reductive treatment with NaBH4 and NaOH. The title cyclic disulfide was then obtained by oxidation of the dithiol (LIX) using oxygen in the presence of FeCl3.
SYN
DE 10036516; WO 0210113
The key dihydroxy ester intermediate (XIII) was also obtained by asymmetric hydrogenation of hydroxy ketoester (XLIII) in the presence of (S)-BINAP-dichlororuthenium catalyst. The precursor hydroxy ketoester (XLIII) was prepared by two alternative procedures. In one method, the racemic dihydroxy ester (XLII) was selectively oxidized to (XLIII) by means of NaOCl. In another method, the unsaturated keto ester (XLIV) was epoxidized by means of sodium percarbonate, and the resultant epoxide (XLV) was then reduced to the hydroxy ketoester (XLIII) by catalytic hydrogenation over PtO2.
SYN
WO 0230919
Both enantiomers of racemic 8-chloro-6-hydroxyoctanoic acid (LX) were separated employing either (+)- or (-)-alpha-methylbenzylamine. Esterification of the (R)-(-)-enantiomer with HCl-MeOH provided the chloro hydroxy ester (LXI). Further chlorination of (LXI) with SOCl2 and pyridine proceeded with inversion of configuration at C-6 to furnish the (S)-dichloro derivative (LXII). The cyclic disulfide (L) was then prepared by treatment of chloride (LXII) with sulfur and sodium sulfide in boiling EtOH. Basic hydrolysis of the methyl ester group of (LXII) then afforded (R) alpha lipoic acid. The title compound was also obtained from the (S)-(+)-acid (LXIII). Reaction of hydroxy acid (LXIII) with methanesulfonyl chloride produced the chloro mesylate (LXIV), which was then cyclized to the target disulfide in the presence of sulfur and Na2S.
SYN
The reaction of the chiral dibenzoyloxy-dihydropyran (LXV) with H2SO4 and HgSO4 gives the unsaturated aldehyde (LXVI), which is condensed with the phosphorane (LXVII) to yield the hepatdienoic ester (LXVIII). The hydrogenation of (LXVIII) with H2 over Pd/C affords the heptanoic ester (LXIX), which is treated with Ts-Cl and pyridine to provide the tosyloxy derivative (LXX). The cyclization of (LXX) by means of K2CO3 gives the chiral epoxide (LXXI), which is condensed with vinylmagnesium bromide (LXXII) to yield 6(S)-hydroxy-8-nonenoic acid methyl ester (LXXIII). The oxidation of the terminal double bond of (LXXIII) with ozone affords the carbaldehyde (LXXIV), which is reduced with NaBH4 to provide 6(S),8-dihydroxyoctanoic acid methyl ester (XLVIII). The reaction of (XLVIII) with Ms-Cl and pyridine gives the dimesylate (XLIX), which is treated with Na2S2 to yield the lipoic acid methyl ester (L), which is hydrolyzed to the target acid with KOH in H2O.
SYN
DE 3629116; EP 0261336
Alkylation of the lithio-dianion of propargyl alcohol (XIII) with 6-bromo-1-hexene (XIV), followed by in situ reduction of the resultant disubstituted acetylene with lithium metal gave the allylic alcohol (XV). Asymmetric Sharpless epoxidation of (XV) using tert-butyl hydroperoxide in the presence of L-(+)-diisopropyl tartrate afforded the (S,S)-epoxy alcohol (XVI). This was reduced to the chiral diol (XVII) employing Red-Al?in THF. After formation of the bis-mesylate (XVIII), oxidative cleavage of the terminal double bond by means of NaIO4 in the presence of ruthenium catalyst furnished the carboxylic acid (XIX). The mesylate groups were finally displaced by sodium disulfide to produce the desired cyclic disulfide compound.
SYN
Both enantiomers of racemic 8-chloro-6-hydroxyoctanoic acid (LX) were separated employing either (+)- or (-)-alpha-methylbenzylamine. Esterification of the (R)-(-)-enantiomer with HCl-MeOH provided the chloro hydroxy ester (LXI). Further chlorination of (LXI) with SOCl2 and pyridine proceeded with inversion of configuration at C-6 to furnish the (S)-dichloro derivative (LXII). The cyclic disulfide (L) was then prepared by treatment of chloride (LXII) with sulfur and sodium sulfide in boiling EtOH. Basic hydrolysis of the methyl ester group of (LXII) then afforded (R) alpha lipoic acid. The title compound was also obtained from the (S)-(+)-acid (LXIII). Reaction of hydroxy acid (LXIII) with methanesulfonyl chloride produced the chloro mesylate (LXIV), which was then cyclized to the target disulfide in the presence of sulfur and Na2S.
DE 19533881; EP 0763533; US 5731448
SYN
WO 9638437
A different strategy was based on the enantioselective oxidation of a cyclohexanone derivative by enzymic Baeyer-Villiger reaction. Keto ester (XXXVIII) was protected as the ethylene ketal (XXXIX) and subsequently reduced to alcohol (XL) using LiAlH4. Acetylation of alcohol (XL) to acetate (XLI), followed by acidic ketal hydrolysis afforded cyclohexanone (XLII) (9,10). The racemic ketone (XLII) was then subjected to oxidative cleavage by monooxigenase 2 obtained from Pseudomonas putida to furnish the (R)-lactone (XLIV) along with unreacted (S)-cyclohexanone (XLIII) (9-11). The use of cyclohexanone monooxigenase from Acinetobacter NCIMB 9871 has also been reported for this reaction (12). Methanolysis of lactone (XLIV) in the presence of NaOMe gave rise to the (R)-dihydroxy ester (XLV). Inversion of the configuration of (XLV) was accomplished by Mitsunobu coupling with p-nitrobenzoic acid (XLVI) to produce the (S)-p-nitrobenzoate ester (XLVII). Smooth hydrolysis of ester (XLVII) provided methyl (S)-6,8-dihydroxyoctanoate (XLVIII), which was processed through intermediates (XLIX) and (L), as for the isopropyl (X) (Scheme 29605101a) and ethyl (XXIX) (Scheme 29605103a) homologues, to afford the title compound.
SYN
Tetrahedron Lett 2001,42(29),4891
The olefinic diester (XXXVIII) was subjected to OsO4-catalyzed asymmetric dihydroxylation using hydroquinidine 1,4-phthalazinediyl diether [(DHQD)2-PHAL] as chiral ligand to afford diol (XXXIX). This was converted to the cyclic sulfate (XL) by treatment with SOCl2, followed by RuCl3-catalyzed NaIO4 oxidation of the intermediate sulfite. Regioselective reduction of sulfate (XL) at the alpha position with NaBH4 in DMA led to the (3S)-alcohol (XLI). Further selective reduction of the ethyl ester group of (XLI) was achieved by treatment with NaBH4-Et3N in MeOH-DMF, yielding the target intermediate dihydroxy ester (XIII).
SYN
1,6-Hexanediol (I) was protected as the mono-tetrahydropyranyl ether (II), and the free hydroxyl group was subsequently oxidized to aldehyde (III) under Swern conditions. Reformatskii reaction of aldehyde (III) with the organozinc reagent generated from ethyl bromoacetate yielded the racemic hydroxy ester (IV). The requisite (S)-enantiomer (VI) was obtained via oxidation of (IV) to oxo ester (V) using pyridinium chlorochromate, and then asymmetric hydrogenation in the presence of (S)-(-)-2,2′-bis(diphenylphosphino)-1,1′-binaphthyl dichlororuthenium complex. Oxo ester (V) was also prepared by SnCl2-catalyzed insertion of ethyl diazoacetate into aldehyde (III). The chiral hydroxy ester (VI) was then reduced to diol (VII) by means of NaBH4-CuSO4. After conversion of (VII) to the corresponding dimesylate (VIII), removal of the tetrahydropyranyl protecting group under acidic conditions gave alcohol (IX). This was sequentially oxidized with PCC to aldehyde, and then with Ag2O to furnish the target dimesylate acid intermediate (X).
SYN
Tetrahedron Asymmetry 2000,11(4),879
The intermediate 6(S)-hydroxy-8-nonenoic acid methyl ester (III) has been obtained by enantioselective allylation of 6-oxohexanoic acid methyl ester (I) with allyltributylstannane (II) catalyzed by the chiral catalyst (R)-BINOL/Ti(O-iPr)4 in refluxing dichloromethane (other BINOL/metal catalysts have also been studied).
SYN
Tetrahedron Lett 1985,26(21),2535
Aldehyde (II), prepared by ozonolysis of cyclohexene (I), was ketalized with (S,S)-2,4-pentanediol (III) to afford dioxane (IV). Titanium chloride-mediated coupling of acetal (IV) with the ketene acetal (V) afforded diastereoselectively adduct (VI), which was subsequently hydrolyzed to carboxylic acid (VII) by means of trifluoroacetic acid. Removal of the pentanediol moiety to furnish the (R)-alcohol (IX) was accomplished via Jones oxidation of the secondary alcohol (VII) to ketone (VIII), followed by beta-elimination in the presence of piperidinium acetate. Reduction of the free carboxyl group by borane-tetrahydrofuran complex gave diol (X), which was further converted to dimesylate (XI). Disulfide displacement of the mesylate groups provided (+)-lipoic acid isopropyl ester (XII), which was finally hydrolyzed to the title acid using K2CO3 in MeOH/H2O.
SYN
Tetrahedron Lett 1987,28(44),5313
A short synthetic strategy utilized the cyclic thioketal (XXXIII), derived from d-menthone (XXXII) and 1,3-propanedithiol, as the chiral template. Stereospecific oxidation of dithiane (XXXIII) employing NaIO4 produced sulfoxide (XXXIV). The carbanion generated from sulfoxide (XXXIV) was stereoselectively alkylated by 5-bromopentanoic acid (XXXV) in the presence of TMEDA to furnish the trans alkylated compound (XXXVI). Finally, acidic hydrolysis of (XXXVI) formed the intermediate mercapto sulfinic acid (XXXVII) which spontaneously cyclized to the desired dithiolane derivative.
SYN
Tetrahedron Lett 1987,28(19),2183
Diisopropylidene mannitol (I) was first converted into the dibutyltin derivative (II), which was subsequently mono-benzylated to (III). Acetylation of (III) with acetic anhydride in pyridine gave (IV). After acidic hydrolysis of the isopropylidene ketals of (IV), the resultant tetraol (V) was converted into tetramesylate (VI). Reductive elimination in (VI) with Zn and NaI produced diene (VII). The acetate group of (VII) was then hydrolyzed to (VIII) using NaOMe. Intermediate (VIII) was reacted with triethyl orthoacetate in the presence of propionic acid to generate the allyl vinyl ether (IX), which underwent a Claisen rearrangement to the diene-ester (X). Selective hydroboration-oxidation of the terminal double bond of (X) yielded the primary alcohol (XI). Subsequent benzyl group hydrogenolysis in (XI) furnished the target intermediate diol (XII).
SYN
Esterification of diisopropylidene mannitol (I) with benzoyl chloride in pyridine afforded dibenzoate (II). Hydrolysis of the isopropylidene ketals of (II) with aqueous HOAc gave tetraol (III), which was further converted to tetramesylate (IV) on treatment with methanesulfonyl chloride and pyridine. Reductive elimination of the mesylate groups of (IV) using Zn dust and NaI yielded diene (V). The benzoate esters of (V) were then removed by treatment with sodium methoxide. The resultant divinylglycol (VI) was reacted with dibutyltin oxide to produce the tin derivative (VII), which was converted to the target intermediate, themono-benzyl ether (VIII), by treatment with benzyl bromide in hot DMF.
SYN
Tetrahedron Lett 1989,30(42),5705
Alkylation of the dianion of octyl acetoacetate (XIII) with 4-iodobutyronitrile (XIV) provided the cyano keto ester (XV). Enantiospecific reduction of (XV) utilizing baker’s yeast gave rise to the desired (S)-hydroxy ester (XVI) in high enantiomeric excess. Subsequent ester group reduction in (XVI) by means of LiBH4 provided diol (XVII). The target dihydroxy ester (XII) was then obtained by alcoholysis of nitrile (XVII) under acidic conditions.
SYN
J Chem Soc Chem Commun 1995,(15),1563
A different strategy was based on the enantioselective oxidation of a cyclohexanone derivative by enzymic Baeyer-Villiger reaction. Keto ester (XXXVIII) was protected as the ethylene ketal (XXXIX) and subsequently reduced to alcohol (XL) using LiAlH4. Acetylation of alcohol (XL) to acetate (XLI), followed by acidic ketal hydrolysis afforded cyclohexanone (XLII) (9,10). The racemic ketone (XLII) was then subjected to oxidative cleavage by monooxigenase 2 obtained from Pseudomonas putida to furnish the (R)-lactone (XLIV) along with unreacted (S)-cyclohexanone (XLIII) (9-11). The use of cyclohexanone monooxigenase from Acinetobacter NCIMB 9871 has also been reported for this reaction (12). Methanolysis of lactone (XLIV) in the presence of NaOMe gave rise to the (R)-dihydroxy ester (XLV). Inversion of the configuration of (XLV) was accomplished by Mitsunobu coupling with p-nitrobenzoic acid (XLVI) to produce the (S)-p-nitrobenzoate ester (XLVII). Smooth hydrolysis of ester (XLVII) provided methyl (S)-6,8-dihydroxyoctanoate (XLVIII), which was processed through intermediates (XLIX) and (L), as for the isopropyl (X) (Scheme 29605101a) and ethyl (XXIX) (Scheme 29605103a) homologues, to afford the title compound.
SYN
Synthesis (Stuttgart) 1996,(5),594
Racemic tetrahydro-2-furylmethanol (I) was converted to tosylate (II), which was further displaced by KCN to yield nitrile (III). Basic hydrolysis of nitrile (III), followed by Fischer esterification of the resultant carboxylic acid (IV) provided ethyl ester (V). Enzymatic resolution of racemic ester (V) by means of the lipase from Candida cylindracea generated a mixture of the (R)-acid (VI) and the unreacted (S)-ester (VII), which were separated by column chromatography. The desired (S) ester (VII) was then reduced to alcohol (VIII) with LiAlH4 in cold Et2O. Regioselective opening of the cyclic ether (VIII) with iodotrimethylsilane in acetone furnished the acetonide of 6-iodo-1,3-hexanediol (IX). Alkylation of benzyl methyl malonate (X) with iodide (IX) provided malonate (XI). Hydrogenolysis of the benzyl ester group of (XI), followed by thermal decarboxylation led to ester (XII). The target dihydroxy ester precursor (XIII) was then obtained by acid-catalyzed hydrolysis of the acetonide function.
SYN
Synthesis (Stuttgart) 1996,(11),1289
Addition of vinylmagnesium bromide to 2-nitrocyclohexanone (XIV) afforded the nitro alcohol (XV). Ring cleavage of (XVI) in the presence of anhydrous CuSO4 absorbed on silica gel gave the nitro ketone (XVI). Nitro group hydrolysis in (XVI) by successive treatment with NaOMe and H2SO4 in MeOH furnished oxo ester (XVII) as the main product. This was enantiospecifically reduced with baker’s yeast to yield the (S)-alcohol (XVIII). Selective methyl ether cleavage with tetrabutylammonium iodide and BF3 provided the dihydroxy ester precursor (XIII).
SYN
An alternative route to (+)-lipoic acid used ethyl 4,6-di-O-acetyl-2,3-dideoxy-alpha-D-erythro-hexopyranoside (XX), prepared from triacetyl-D-glucal, as the chiral starting point. Deacetylation of (XX) with sodium methoxide under Zemplen conditions gave diol (XXI) which, after conventional benzylation, led to the 4,6-di-O-benzyl derivative (XXII). Ring opening of the cyclic acetal (XXII) with propanediol in the presence of boron trifluoride afforded the dithiane derivative (XXIII). The free hydroxyl group of (XXIII) was converted into xanthate (XXIV) by reaction with NaH and CS2, followed by methyl iodide. Reductive cleavage of the xanthate group by means of Bu3SnH and AIBN provided (XXV). Hydrolysis of the thioacetal function with HgO and BF3 provided aldehyde (XXVI). Chain homologation was performed by Wittig reaction of aldehyde (XXVI) with phosphorane (XXVII) to afford the unsaturated ester (XXVIII). Simultaneous double bond hydrogenation and benzyl ether cleavage in the presence of Raney nickel led to dihydroxy ester (XXIX). This was converted to the corresponding dimesylate (XXX), which was further cyclized to disulfide (XXXI) using the in situ generated sodium disulfide as in the precedent Schemes. Finally, basic hydrolysis of the ethyl ester (XXXI) yielded the title carboxylic acid.
Carbohydr Res 1986,148(1),51
SYN
Diisopropylidene mannitol (I) was first converted into the dibutyltin derivative (II), which was subsequently mono-benzylated to (III). Acetylation of (III) with acetic anhydride in pyridine gave (IV). After acidic hydrolysis of the isopropylidene ketals of (IV), the resultant tetraol (V) was converted into tetramesylate (VI). Reductive elimination in (VI) with Zn and NaI produced diene (VII). The acetate group of (VII) was then hydrolyzed to (VIII) using NaOMe. Intermediate (VIII) was reacted with triethyl orthoacetate in the presence of propionic acid to generate the allyl vinyl ether (IX), which underwent a Claisen rearrangement to the diene-ester (X). Selective hydroboration-oxidation of the terminal double bond of (X) yielded the primary alcohol (XI). Subsequent benzyl group hydrogenolysis in (XI) furnished the target intermediate diol (XII).
^ Reljanovic, M; Reichel, G; Rett, K; Lobisch, M; et al. (September 1999). “Treatment of diabetic polyneuropathy with the antioxidant thioctic acid (alpha-lipoic acid): A two year multicenter randomized double-blind placebo-controlled trial (ALADIN II). Alpha Lipoic Acid in Diabetic Neuropathy”. Free Radical Research. 31 (3): 171–9. doi:10.1080/10715769900300721. PMID10499773.
^ Durrani, AI; Schwartz, H; Nagl, M; Sontag, G (October 2010). “Determination of free [alpha]-lipoic acid in foodstuffs by HPLC coupled with CEAD and ESI-MS”. Food Chemistry. 120 (4): 38329–36. doi:10.1016/j.foodchem.2009.11.045.
^ Teichert, J; Preiss, R (November 1992). “HPLC-methods for determination of lipoic acid and its reduced form in human plasma”. International Journal of Clinical Pharmacology, Therapy, and Toxicology. 30 (11): 511–2. PMID1490813.
^ Biewenga, GP; Haenen, GR; Bast, A (September 1997). “The pharmacology of the antioxidant lipoic acid”. General Pharmacology. 29 (3): 315–31. doi:10.1016/S0306-3623(96)00474-0. PMID9378235.
^ Jump up to:ab Kleeman, A; Borbe, HO; Ulrich, H (1991). “Thioctic Acid-Lipoic Acid”. In Borbe, HO; Ulrich, H (eds.). Thioctsäure: Neue Biochemische, Pharmakologische und Klinische Erkenntnisse zur Thioctsäure [Thioctic Acid. New Biochemistry, Pharmacology and Findings from Clinical Practice with Thioctic Acid]. Symposium at Wiesbaden, DE, 16–18 February 1989. Frankfurt, DE: Verlag. pp. 11–26. ISBN9783891191255.
^ Fontanella, L (1955). “Preparation of optical antipodes of alpha-lipoic acid”. Il Farmaco; Edizione Scientifica. 10 (12): 1043–5. PMID13294188.
^ Lang, G (1992). In Vitro Metabolism of a-Lipoic Acid Especially Taking Enantioselective Bio-transformation into Account (Ph.D. thesis). Münster, DE: University of Münster.
^US patent 5281722, Blaschke, G; Scheidmantel, U & Bethge, H et al., “Preparation and use of salts of the pure enantiomers of alpha-lipoic acid”, issued 1994-01-25, assigned to DeGussa.
^ Jump up to:ab Carlson, DA; Young, KL; Fischer, SJ; Ulrich, H. “Ch. 10: An Evaluation of the Stability and Pharmacokinetics of R-lipoic Acid and R-Dihydrolipoic Acid Dosage Forms in Plasma from Healthy Human Subjects”. Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects. pp. 235–70. In Packer & Patel 2008.
^ Packer, L; Kraemer, K; Rimbach, G (October 2001). “Molecular aspects of lipoic acid in the prevention of diabetes complications”. Nutrition. 17 (10): 888–95. doi:10.1016/S0899-9007(01)00658-X. PMID11684397.
^ Hill, AS; Werner, JA; Rogers, QR; O’Neill, SL; et al. (April 2004). “Lipoic acid is 10 times more toxic in cats than reported in humans, dogs or rats”. Journal of Animal Physiology and Animal Nutrition. 88 (3–4): 150–6. doi:10.1111/j.1439-0396.2003.00472.x. PMID15059240.
^ Jump up to:ab Shay, KP; Shenvi, S; Hagen, TM. “Ch. 14 Lipoic Acid as an Inducer of Phase II Detoxification Enzymes Through Activation of Nr-f2 Dependent Gene Expression”. Lipoic Acid: Energy Production, Antioxidant Activity and Health Effects. pp. 349–71. In Packer & Patel 2008.
^ Biewenga, GP; Haenen, GRMM; Bast, A (1997). “Ch. 1: An Overview of Lipoate Chemistry”. In Fuchs, J; Packer, L; Zimmer, G (eds.). Lipoic Acid In Health & Disease. CRC Press. pp. 1–32. ISBN9780824700935.
^US patent 6271254, Ulrich, H; Weischer, CH & Engel, J et al., “Pharmaceutical compositions containing R-alpha-lipoic acid or S-alpha.-lipoic acid as active ingredient”, issued 2001-08-07, assigned to ASTA Pharma.
^ Kilic, F; Handelman, GJ; Serbinova, E; Packer, L; et al. (October 1995). “Modelling cortical cataractogenesis 17: In vitro effect of a-lipoic acid on glucose-induced lens membrane damage, a model of diabetic cataractogenesis”. Biochemistry and Molecular Biology International. 37 (2): 361–70. PMID8673020.
^ Artwohl, M; Schmetterer, L; Rainer, G; et al. (September 2000). Modulation by antioxidants of endothelial apoptosis, proliferation, & associated gene/protein expression. 36th Annual Meeting of the European Association for the Study of Diabetes, 17–21 September 2000, Jerusalem, Israel. Diabetologia. 43 (Suppl 1) (published August 2000). Abs 274. PMID11008622.
^ Frölich, L; Götz, ME; Weinmüller, M; Youdim, MB; et al. (March 2004). “(r)-, but not (s)-alpha lipoic acid stimulates deficient brain pyruvate dehydrogenase complex in vascular dementia, but not in Alzheimer dementia”. Journal of Neural Transmission. 111 (3): 295–310. doi:10.1007/s00702-003-0043-5. PMID14991456. S2CID20214857.
^ Ziegle, D.; Reljanovic, M; Mehnert, H; Gries, F. A. (1999). “α-Lipoic acid in the treatment of diabetic polyneuropathy in Germany”. Experimental and Clinical Endocrinology & Diabetes. 107 (7): 421–30. doi:10.1055/s-0029-1212132. PMID10595592.
6,10-Epoxy-6H,16H-diindolo[1,2,3-gh:3′,2′,1′-lm]pyrrolo[3,4-j][1,7]benzodiazonin-16-one, 7,8,9,10,17,18-hexahydro-7-methoxy-6-methyl-8-(methylamino)-, (6S,7R,8R,10R)- 62996-74-1[RN] AM-2282 Antibiotic 230 antibiotic am 2282 StaurosporineCAS Registry Number: 62996-74-1 CAS Name: (9S,10R,11R,13R)- 2,3,10,11,12,13-Hexahydro-10-methoxy-9-methyl-11-(methylamino)-9,13-epoxy-1H,9H-diindolo[1,2,3-gh:3¢,2¢,1¢-lm]pyrrolo[3,4-j][1,7]benzodiazonin-1-one Manufacturers’ Codes: AM-2282; CGP-39360 Molecular Formula: C28H26N4O3, Molecular Weight: 466.53 Percent Composition: C 72.09%, H 5.62%, N 12.01%, O 10.29% Literature References: Protein kinase C inhibitor; alkaloid isolated from Streptomyces staurosporeus. Isoln: S. Omura et al., J. Antibiot.30, 275 (1977). Crystal and molecular structure: A. Furusaki et al., J. Chem. Soc. Chem. Commun.1978, 800; eidem,Bull. Chem. Soc. Jpn.55, 3681 (1982). Corrected stereochemistry: N. Funato et al., Tetrahedron Lett.35, 1251 (1994). Total synthesis: J. T. Link et al., J. Am. Chem. Soc.117, 552 (1995); idem et al., ibid.118, 2825 (1996). Biosynthetic studies: D. Meksuriyen, G. A. Cordell, J. Nat. Prod.51, 884, 893 (1988); S.-W. Yang et al., ibid.62 1551 (1999). HPLC determn in blood and pharmacokinetics in rats: L. R. Gurley et al., J. Chromatogr. B712, 211 (1998). Inhibition of protein kinase C: T. Tamaoki et al., Biochem. Biophys. Res. Commun.135, 397 (1986); of other protein kinases: U. T. Rüegg, G. M. Burgess, Trends Pharmacol. Sci.10, 218 (1989). Induction of apoptosis: E. Falcieri et al., Biochem. Biophys. Res. Commun.193, 19 (1993); R. Bertrand et al., Exp. Cell Res.211, 314 (1994); of tyrosine phosphorylation: D. Rasouly, P. Lazarovici, Eur. J. Pharmacol.269, 255 (1994). Properties: Pale yellow needles from chloroform-methanol as the methanol solvate, mp 270° (dec) (Omura). Also reported as yellow crystals from methanol, mp 288-291° (Meksuriyen, Cordell). [a]D25 +35.0° (c = 1 in methanol); [a]D22 +56.1° (c = 0.14 in methanol). uv max (methanol): 241.0, 266.0, 292.5, 321.5, 335.0, 355.0, 372.5 nm (log e 4.25, 4.26, 4.53, 3.88, 3.96, 3.81, 3.85). Sol in DMSO, DMF. Slightly sol in chloroform, methanol. Melting point: mp 270° (dec); mp 288-291° (Meksuriyen, Cordell) Optical Rotation: [a]D25 +35.0° (c = 1 in methanol); [a]D22 +56.1° (c = 0.14 in methanol) Absorption maximum: uv max (methanol): 241.0, 266.0, 292.5, 321.5, 335.0, 355.0, 372.5 nm (log e 4.25, 4.26, 4.53, 3.88, 3.96, 3.81, 3.85) Derivative Type: Hydrochloride Molecular Formula: C28H26N4O3.HCl, Molecular Weight: 502.99 Percent Composition: C 66.86%, H 5.41%, N 11.14%, O 9.54%, Cl 7.05% Properties: LD50 in mice (mg/kg): 6.6 i.p. (Omura). Toxicity data: LD50 in mice (mg/kg): 6.6 i.p. (Omura) Use: Pharmacological tool to study signal transduction pathways, tyrosine phosphorylation and to induce apoptosis. An indolocarbazole that is a potent protein kinase C inhibitor which enhances cAMP-mediated responses in human neuroblastoma cells. (Biochem Biophys Res Commun 1995;214(3):1114-20)
Staurosporine (antibiotic AM-2282 or STS) is a natural product originally isolated in 1977 from the bacterium Streptomyces staurosporeus.[1] It was the first of over 50 alkaloids to be isolated with this type of bis-indole chemical structure. The chemical structure of staurosporine was elucidated by X-ray analysis of a single crystal and the absolute stereochemical configuration by the same method in 1994.[2]
Staurosporine was discovered to have biological activities ranging from anti-fungal to anti-hypertensive.[3] The interest in these activities resulted in a large investigative effort in chemistry and biology and the discovery of the potential for anti-cancer treatment.
|00501 ] As will become apparent to a skilled artisan, many of the bridged epoxy diindolopyrrolo-hexahydrobenzodiazocines are commercially available as final compounds or modifiable intermediates. Staurosporine was originally isolated from the bacterium Streptomyces staurosporeus. (S.Omura et al. J.Antibiotics, 30, 275
1977).
[00502] Synthesis of 9, 12-epoxy staurosporine analogs:
NOTE: 2) 120°, 6) 120°, Reactantsi 4, Reagents : 7, Catalysts. 2, Solvents 18, Steps: 9, Stages: 11, Most stages in any one stept 2
[00503] Greater detail is provided in Tetrahedron Letters, 36(46), 8383-6,
1995. [00504] Alternative synthesis of 9,12-epoxy staurosporine analogs:
NOTEt 1) stereoselective, 5) Raney nickel present, Reactantsi 5, Reagentsi 6, Catalystsi 4, Solvents! 5, Stepsi 7, stagest 9, Most stages in any one step: 2 [00505] Greater detail is provided in Organic Letters, 3(11), 1689-1692; 2001. [00506] [00507] Synthesis of 9, 13-epoxy staurosporine analogs:
[00508] Whereas, a more thorough description of reagents, reaction conditions, and other pertinent syntheses are described Journal of the American Chemical Society, 117(1), 552-3; 1995. Additionally, syntheses on staurosporine and analogs thereof are described by S.J Danishefsky et al, J.Am.Chem.Soc, 118, 28251996 and J.L.Wood et al, J.Am.Chem.Soc, 118, 106561996.
Example 1Method for obtaining crude midostaurin B1 from crude staurosporine A1
A reactor was loaded with crude staurosporine A1 (1 mol) and DMF (7 L). The solution was cooled to 0°C and subsequently DIPEA (1.5 mol) was added. Benzoyl chloride (1.2 mol) was added while keeping the temperature within the range 0-5°C. After 30 minutes from the end of the addition, an aqueous 1 % ammonium chloride solution (15 L) was added while keeping the temperature within the range 0-5°C. After 1 hour from the end of the addition, the suspension was filtered and the panel was washed with plenty of water. The solid was dried for 6 hours at 40°C, obtaining crude midostaurin B1 with 95% yield. Example 2Method for obtaining purified midostaurin B2 from crude midostaurin B1 – reduction of 3-hydroxymidostaurin to midostaurin with triethylsilanei. TFA/TESii. NaHC03iii. Crystallizationin MeTHFiv. Crystallization
in EtOH/H20
B4 B2
A reactor was loaded with crude midostaurin B1 (1 mol) and DCM (10 L). The solution was cooled to 0°C and subsequently added with TES (1 mol) and TFA (0.50 L) in this order, while keeping the temperature within the range 0-5°C. At the end of the additions the solution was brought to 20°C. After 3 hours the solution was added with an aqueous 5% sodium bicarbonate solution (20 L). At the end of the development of gas the resulting two phases were separated and the aqueous phase was washed twice with DCM (10 L). The collected organic phases were concentrated at atmospheric pressure, added with 2-MeTHF (30 L) and two changes of solvent at atmospheric pressure were carried out. The solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C and subsequently transferred into another reactor. Ethanol (7 L) was added and the mixture was heated at 75°C up to complete dissolution. Water (30 L) was added with a concurrent cooling to 20°C. The resulting suspension was filtered and the panel was washed with plenty of water. The solid was dried for 12 hours at 80°C, obtaining purified midostaurin B2 with 85% yield. Example 3Method for obtaining purified staurosporine A2 from crude staurosporine A1 – reduction of 3-hydroxystaurosporine to staurosporine with triethylsilane
A reactor was loaded with crude staurosporine A1 (1 mol) and DCM (10 L). The solution was cooled to 0°C and subsequently added with TES (1 mol) and TFA (0.50 L) in this order, while keeping the temperature within the range 0-5°C. After 1 hour from the end of the additions, the solution was added with MeOH (10 L) and, subsequently, with an aqueous 5% sodium bicarbonate solution (20 L). At the end of the development of gas the resulting two phases were separated and the aqueous phase was washed twice with DCM (10 L). The collected organic phases were concentrated at atmospheric pressure, added with 2-MeTHF (50 L) and two changes of solvent at atmospheric pressure were carried out. The warm solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C, obtaining purified staurosporine A2 with 80% yield. Example 4Method for obtaining purified staurosporine A2 from crude staurosporine A1 – derivatization of 3-hydroxystaurosporine with trifluoroacetic acid and purification by crystallization
A reactor was loaded with crude staurosporine A1 (1 mol) and DCM (10 L). The mixture was cooled to 0°C and added with TFA (0.50 L), while keeping the temperature within the range 0-5°C. After 1 hour from the end of the addition, the solution was added with MeOH (10 L) and, subsequently, with an aqueous 5% sodium bicarbonate solution (20 L). At the end of the development of gas the resulting two phases were separated and the aqueous phase was washed twice with DCM (10 L). The collected organic phases were concentrated at atmospheric pressure, added with 2-MeTHF (50 L) and two changes of solvent at atmospheric pressure were carried out. The warm solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C, obtaining purified staurosporine A2 with 80% yield. Example 5Method for obtaining purified midostaurin B2 from purified staurosporine A2 i. BzCI/DIPEAii. NH4CI/H2Oiii. Crystallizationin MeTHFiv. Crystallization
in EtOH/H20
A2 B2
A reactor was loaded with purified staurosporine A2 (1 mol) and DMF (7 L). The solution was cooled to 0°C and subsequently DIPEA (1.5 mol) was added. Benzoyl chloride (1.2 mol) was added while keeping the temperature within the range 0-5°C. After 30 minutes from the end of the addition, an aqueous 1 % ammonium chloride solution (15 L) was added while keeping the temperature within the range 0-5°C. After 1 hour from the end of the addition, the suspension was filtered and the panel was washed with plenty of water. The solid was dried for 6 hours at 40°C and subsequently transferred into another reactor. 2-MeTHF (30 L) was added and the suspension was heated under reflux up to complete dissolution. The solution was clarified by filtration at 75°C and the panel was washed with 2-MeTHF. The filtrate was transferred into another reactor and cooled at 0°C in 8 hours. After further 2 hours at 0°C the suspension was filtered and the panel was washed twice with 2-MeTHF. The solid was dried for 12 hours at 80°C and subsequently transferred into another reactor. Ethanol (7 L) was added and the mixture was heated at 75°C up to complete dissolution. Water (30 L) was added with a concurrent cooling to 20°C. The resulting suspension was filtered and the panel was washed with plenty of water. The solid was dried for 12 hours at 80°C, obtaining purified midostaurin B2 with 85% yield. ClaimsHide Dependent 1) A process for the preparation of midostaurin with high purity, that is with a content of 3-hydroxymidostaurin impurities (III) and (IV) lower than 0.1%, comprising the treatment with strong organic or inorganic acids in a water-immiscible solvent and, optionally, also with reducing silanes.2) The process for the preparation of midostaurin according to claim 1 , comprising the treatment of crude midostaurin with a reducing silane in the presence of a strong organic or inorganic acid.3) The process for the preparation of midostaurin according to claim 1 , comprising the treatment of crude staurosporine with a strong organic or inorganic acid, optionally with the concomitant addition of a reducing silane.4) The process for the preparation of midostaurin according to claim 1 , 2 or 3, wherein the water-immiscible solvent is an aprotic polar water-immiscible solvent.5) The process for the preparation of midostaurin according to claim 4 wherein the water-immiscible solvent is dichloromethane, dichloroethane, methyl tetrahydrofuran or methylethylketone, preferably dichloromethane.6) The process for the preparation of midostaurin according to claim 1 , 2 or 3, wherein the strong acid is trifluoroacetic acid.7) The process for the preparation of midostaurin according to claim 1 , 2 or 3, wherein the reducing silane is triethylsilane.8) The process for the preparation of midostaturin according to anyone of the preceding claims, further comprising the benzoylation reaction of staurosporine to midostaurin characterized in that the benzoylation reaction is quenched with an aqueous solution having a slightly acid pH.9) The process for the preparation of midostaurin according to claim 8 wherein the aqueous solution having a slightly acid pH is an aqueous ammonium chloride solution.10) The process for the preparation of midostaurin according to anyone of the preceding claims, comprising the obtainment of purified midostaurin by crystallization from 2-MeTHF and its further isolation by:dissolving the crystallized midostaurin in a water-miscible polar solvent, adding waterisolating purified midostaurin as an amorphous solid obtained by filtering and drying, with a content of organic solvents < 50ppm. 11) The process for the preparation of purified midostaurin according to claim 10, wherein the polar s
Patent
Publication numberPriority datePublication dateAssigneeTitleJPS5247055B21973-12-041977-11-30US5093330A1987-06-151992-03-03Ciba-Geigy CorporationStaurosporine derivatives substituted at methylamino nitrogenEP0575955A11992-06-221993-12-29Kyowa Hakko Kogyo Co., Ltd.Process for producing staurosporine derivativesWO2006048296A12004-11-052006-05-11Novartis AgOrganic compoundsWO2011064355A12009-11-302011-06-03Novartis AgPolymorphous forms iii and iv of n-benzoyl staurosporineWO2018165071A12017-03-062018-09-13Teva Pharmaceutical Works Ltd.Solid state forms of midostaurin
Biological activities
The main biological activity of staurosporine is the inhibition of protein kinases through the prevention of ATP binding to the kinase. This is achieved through the stronger affinity of staurosporine to the ATP-binding site on the kinase. Staurosporine is a prototypical ATP-competitive kinase inhibitor in that it binds to many kinases with high affinity, though with little selectivity.[4] Structural analysis of kinase pockets demonstrated that main chain atoms which are conserved in their relative positions to staurosporine contributes to staurosporine promiscuity.[5] This lack of specificity has precluded its clinical use, but has made it a valuable research tool. In research, staurosporine is used to induce apoptosis. The mechanism of how it mediates this is not well understood. It has been found that one way in which staurosporine induces apoptosis is by activating caspase-3.[6] At lower concentration, depending on the cell type, staurosporine induces specific cell cycle effects arresting cells either in G1 or in G2 phase of the cell cycle.[7]
Staurosporine is an indolocarbazole. It belongs to the most frequently isolated group of indolocarbazoles: Indolo(2,3-a)carbazoles. Of these, Staurosporine falls within the most common subgroup, called Indolo(2,3-a)pyrrole(3,4-c)carbazoles. These fall into two classes – halogenated (chlorinated) and non-halogenated. Halogenated indolo(2,3-a)pyrrole(3,4-c)carbazoles have a fully oxidized C-7 carbon with only one indole nitrogen containing a β-glycosidic bond, while non-halogenated indolo(2,3-a)pyrrole(3,4-c)carbazoles have both indole nitrogens glycosylated, and a fully reduced C-7 carbon. Staurosporine is in the non-halogenated class.[8]
Staurosporine is the precursor of the novel protein kinase inhibitormidostaurin (PKC412).[9][10] Besides midostaurin, staurosporine is also used as a starting material in the commercial synthesis of K252c (also called staurosporine aglycone). In the natural biosynthetic pathway, K252c is a precursor of staurosporine.
Structure of an Indolo[2,3-a]pyrrole[3,4-c]carbazol
Biosynthesis
The biosynthesis of staurosporine starts with the amino acid L-tryptophan in its zwitterionic form. Tryptophan is converted to an imine by enzyme StaO which is an L-amino acid oxidase (that may be FAD dependent). The imine is acted upon by StaD to form an uncharacterized intermediate proposed to be the dimerization product between 2 imine molecules. Chromopyrrolic acid is the molecule formed from this intermediate after the loss of VioE (used in the biosynthesis of violacein – a natural product formed from a branch point in this pathway that also diverges to form rebeccamycin. An aryl aryl coupling thought to be catalyzed by a cytochrome P450 enzyme to form an aromatic ring system occurs.[8]
This is followed by a nucleophilic attack between the indole nitrogens resulting in cyclization and then decarboxylation assisted by StaC exclusively forming staurosporine aglycone or K252c. Glucose is transformed to NTP-L-ristoamine by StaA/B/E/J/I/K which is then added on to the staurosporine aglycone at 1 indole N by StaG. The StaN enzyme reorients the sugar by attaching it to the 2nd indole nitrogen into an unfavored conformation to form intermediated O-demethyl-N-demethyl-staurosporine. Lastly, O-methylation of the 4’amine by StaMA and N-methylation of the 3′-hydroxy by StaMB leads to the formation of staurosporine.[8]
Research in clinical use
When encapsulated in liposomenanoparticle, staurosporine is shown to suppress tumors in vivo in a mouse model without the toxic side effects which have prohibited its use as an anti-cancer drug with high apoptotic activity. Researchers in UC San Diego Moores Cancer Center develop a platform technology of high drug-loading efficiency by manipulating the pH environment of the cells. When injected into the mouse glioblastoma model, staurosporine is found to accumulate primarily in the tumor via fluorescence confirmation, and the mice did not suffer weight loss compared to the control mice administered with the free compound, an indicator of reduced toxicity.[11][12]
^ Funato N, Takayanagi H, Konda Y, Toda Y, Harigaya Y, Omura S (1994). “Absolute configuration of staurosporine by X-ray analysis”. Tetrahedron Lett. 35 (8): 1251–1254. doi:10.1016/0040-4039(94)88036-0.
^[1] Rüegg UT, Burgess GM. (1989) Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends in Pharmacological Science 10 (6): 218-220.
^ Chae HJ, Kang JS, Byun JO, Han KS, Kim DU, Oh SM, Kim HM, Chae SW, Kim HR (2000). “Molecular mechanism of staurosporine-induced apoptosis in osteoblasts”. Pharmacological Research. 42 (4): 373–381. doi:10.1006/phrs.2000.0700. PMID10987998.
^ Bruno S, Ardelt B, Skierski JS, Traganos F, Darzynkiewicz Z (1992). “Different effects of staurosporine, an inhibitor of protein kinases, on the cell cycle and chromatin structure of normal and leukemic lymphocytes”. Cancer Res. 52 (2): 470–473. PMID1728418.
Marbofloxacin is a carboxylic acid derivative third generation fluoroquinoloneantibiotic. It is used in veterinary medicine under the trade names Marbocyl, Forcyl, Marbo vet and Zeniquin. A formulation of marbofloxacin combined with clotrimazole and dexamethasone is available under the name Aurizon (CAS number 115550-35-1).
https://patents.google.com/patent/CN107383058B/enMarbofloxacin (Marbofloxacin) is fluoroquinolone antibacterial agent for animals, the entitled fluoro- 3- methyl-1 0- of 9- of chemistry (4- methylpiperazine-1-yl) -7- oxo -2,3- dihydro -7H- pyridine [3,2,1-ij] [4,1,2] benzo oxadiazines -6- carboxylic acid, It is developed by Roche Holding Ag, and is further developed by French Vetoquinol (method national strength and prestige are grand) company earliest, in nineteen ninety-five in Europe Listing.Marbofloxacin is after Enrofloxacin (Enrofloxacin), Danofloxacin (Danofloxacin), sarafloxacin (Sarafloxacin) etc. another third generation carbostyril family antibacterial drugs after, the drug have extensive antibacterial activity simultaneously With very good dynamic characteristic, sterilizing power is strong, absorbs fastly, widely distributed in vivo, with other antimicrobials without crossing drug resistant Property, easy to use, adverse reaction is small.Pharmacokinetic is studies have shown that Marbofloxacin removes long half time in animal body, biology Availability, almost without residual in the blood of animal, excrement and tissue, is well suited for clinically to antibiosis for animals close to 100% The requirement of element, structural formula are as follows: Structure is complicated for Marbofloxacin, not only contains methyl piperazine substituent group, but also aromatic moieties contain pyridine benzo evil two Piperazine skeleton has had many documents and patent report at present and has reviewed its synthetic method, such as patent US4801584, ZL94190968.9, EP2010/067828, CN101619068, CN102060860, CN102617595, document J.Org. Chem., 1992,57 (2), 744-766, ” chemical reagent ” 2007,29 (11), 701-703., ” Chinese Journal of Pharmaceuticals ” 2002,33 (1), 1358-1363 etc..Patent US4801584 reports fluoro- via the fluoro- 4,8- dihydroquinoline -3- carboxylic acid, ethyl ester of 6,7- bis- preparation 6,7- bis- The method of 8- hydroxyl -1- (methylamino) -4- oxo-Isosorbide-5-Nitrae-dihydroquinoline -3- carboxylic acid, ethyl ester, this method are related to using valuableness And commercialization is not easy amination reagent O- (2, the 4- dinitrophenyl) oxyammonia largely purchased in 1 upper amino, by multistep reaction After complete the preparation of fluoro- 8- hydroxyl -1- (the methylamino) -4- oxo-Isosorbide-5-Nitrae-dihydroquinoline -3- carboxylic acid, ethyl ester of 6,7- bis-, passed through after It crosses and paraformaldehyde, N methyl piperazine reacts the preparation for realizing Marbofloxacin.Correlated response formula is as follows: The patent literature reports such as patent ZL94190968.9 are that raw material prepares Ma Bosha from 2,3,4,5 tetra fluoro benzoic acid The synthetic route of star, this method are not only related to the multisteps hazardous reactions such as carboxylic acyloxy chlorination, Grignard Reagent preparation reaction, synthesize road Wire length, and 3- (the N- methyl formyl hydrazono-) ethyl acrylate for being difficult to prepare is used, and yield is low, be not suitable for industrially putting Mass production, correlated response formula are as follows: Patent CN101619068 is condensed using 2,3,4,5- phenyl tetrafluoride carbamoylalkyl esters and inferior amine salt, obtained N- bis- Methyl substituted enamine derivates react the enamine for preparing the substitution of N- methyl-N- acyl group under organic acid catalysis with N- methylhydrazide Derivative, then 6,7,8- tri- fluoro- 1- (methylamino) -4- oxo-Isosorbide-5-Nitrae-dihydroquinoline-are completed in cyclization and hydrolysis under alkaline condition The preparation of 3- carboxylic acid realizes Ma Bosha finally by with N methyl piperazine, dimethyl formal (or diethyl formal) reaction The preparation of star.The technique uses the dimethyl suflfate and the height hazardous reaction reagent such as sodium hydride or alkalide of severe toxicity, because And it is subject to certain restrictions in commercial process.Correlated response formula is as follows: In conclusion there are various deficiencies, such as chemistry examinations in the synthetic route of existing synthesis Marbofloxacin The defects of agent is expensive, reaction route is too long, using the chemical reagent for being unfavorable for industrialized production, the present inventor are real after study It tests, invents a kind of new method for preparing Marbofloxacin.The preparation of embodiment 1:1,1,1- tri- chloro- 4- (4- methylpiperazine-1-yl) butyl- 3- alkene -2- ketone(E) -1,1,1- tri- chloro-4-methoxy butyl- 3- alkene -2- ketone (Formulas I, R=Me) (10.18g, 50mmol), 1- methyl The mixture of piperazine (6.0g, 60mmol) and mesitylene (50mL) is heated to reflux temperature and stirs 6 hours, and system is natural Be cooled to room temperature, remove organic solvent under high vacuum reduced pressure, residue (14.2g, crude product do not purify) without further purification, directly It connects for reacting in next step.Embodiment 2:(6,8- bis- fluoro- 7- (4- methylpiperazine-1-yl) -4- oxo -3- (2,2,2- trichloroacetyl) quinoline Quinoline -1 (4H)-yl) urethanes (Formula VII) preparationUnder nitrogen protection, the product (14.2g is not purified, is directly used) of embodiment 1 is dissolved in toluene (120mL), then body Triethylamine (72mL, 514mmol) is added in system, system is heated to reflux temperature.Under reflux temperature, slowly dripped into reaction system Add toluene (60mL) solution of 2,3,4,5- phenyl tetrafluoride formyl chloride (16g, 75.3mmol).Rear system reflux is added dropwise 30min, then system slow cooling is to 60 DEG C, heat filtering.Filtrate is transferred in 500ml reaction flask, and carbazic acid second is then added Ester (Formula V, R2=Et) (6.25g, 60mmol).System is reacted 12 hours at a temperature of 60-65 DEG C after addition.To reaction H is slowly added in system2O (150mL) quenching reaction, system are naturally cooling to room temperature.Filtering, obtains solid, and solid uses heptan Alkane/ethyl acetate system mashing processing, obtains solid (Formula VII, R2=Et) (21.2g).Embodiment 3:1- amino -6- fluoro- 8- hydroxyl -7- (4- methylpiperazine-1-yl) -4- oxo -1,4- dihydroquinoline -3- The preparation of carboxylic acid (Formula VIII)2 obtained solid of embodiment (21.2g) is placed in 200ml reaction flask, ethyl alcohol (50mL) is added into reaction system With water (50mL), system is heated to flowing back.The aqueous solution (30mL) of KOH (7.0g) is slowly added under counterflow condition to system, is dripped System maintains the reflux for state response 96 hours after adding.System is naturally cooling to room temperature, and H is added in system2O (100mL) and CH2Cl2(50ml) stands after stirring and separates organic phase, and water phase reuses CH2Cl2It is extracted twice (2 × 50mL).Water phase uses salt Sour regulation system is to acid (pH=3-4), and then water phase reuses CH2Cl2It is extracted twice (2 × 100mL), merges organic phase, subtract Pressure-off obtains solid (Formula VIII) (12.4g) after removing organic solvent.The preparation of embodiment 4:1,1,1- tri- chloro- 4- (4- methylpiperazine-1-yl) butyl- 3- alkene -2- ketoneSequentially added in reaction flask the chloro- 4- ethyoxyl butyl- 3- alkene -2- ketone (Formulas I, R=Et) of (E) -1,1,1- three (14.1g, 65mmol) and 1- methyl piperazine (7.0g, 70mmol).Then system is heated to 130-155 DEG C and is stirred to react 5 hours.System is cold But to room temperature, the complete raw material of a little unreacted of high vacuum removed under reduced pressure, residue (16.8g, crude product do not purify) is without pure Change, is directly used in and reacts in next step.Embodiment 5:(6,8- bis- fluoro- 7- (4- methylpiperazine-1-yl) -4- oxo -3- (2,2,2- trichloroacetyl) quinoline Quinoline -1 (4H)-yl) t-butyl carbamate (Formula VII, R2=tBu) preparationUnder nitrogen protection, the product (16.0g is not purified, is directly used) of embodiment 4 is dissolved in toluene (125mL), then N is added in system, N- diisopropylethylamine (104.5mL, 600mmol), system is heated to reflux temperature.Under reflux temperature, to Toluene (70mL) solution of 2,3,4,5- phenyl tetrafluoride formyl chloride (18.8g, 88mmol) is slowly added dropwise in reaction system.It is added dropwise Starting material Formula II is tracked to HPLC within system reflux 1 hour afterwards to disappear.Then system slow cooling is to 60 DEG C or so, hot mistake Filter.Filtrate is transferred in 500mL reaction flask, and tert-butyl carbazate (Formula V, R is then added2=tBu)(9.3g,70mmol).It is added After system reacted 48 hours at a temperature of 60-65 DEG C.H is slowly added into reaction system2O (150mL) quenching reaction, body System is naturally cooling to room temperature.Filtering obtains solid, and solid is handled using heptane/ethyl acetate system mashing, obtains solid (formula VII,R2=tBu) (19.3g) is directly used in next step without further purification.Embodiment 6:1- amino -6- fluoro- 8- hydroxyl -7- (4- methylpiperazine-1-yl) -4- oxo -1,4- dihydroquinoline -3- The preparation of carboxylic acid (Formula VIII)By 5 obtained solid of embodiment (19.0g) as in 200mL reaction flask, methanol (55mL) is added into reaction system With water (55mL), system is heated to flowing back.The aqueous solution (30mL) of CsOH (13.5g) is slowly added under counterflow condition to system, Rear system is added dropwise and maintains the reflux for state response 96 hours.System is naturally cooling to room temperature, and H is added in system2O (100mL) and CH2Cl2(50mL) stands after stirring and separates organic phase, and water phase reuses CH2Cl2It is extracted twice (2 × 50mL).Water phase uses salt Sour regulation system is to acid (pH=3-4), and then water phase reuses CH2Cl2It is extracted twice (2 × 100mL), merges organic phase, subtract Pressure-off obtains solid (Formula VIII) (8.8g) after removing organic solvent.Embodiment 7: the preparation of Marbofloxacin1- amino-6- fluoro- 8- hydroxyl-7- (4- methylpiperazine-1-yl) oxo-1-4- is sequentially added in 100mL reaction flask, 4- dihydroquinoline -3- carboxylic acid (Formula VIII, 6.0g), 85% formic acid (30mL) and 36.5% formalin (6.0mL). System is carefully slowly heated to 75 DEG C or so reactions 1 hour after addition.Then system is cooled to 10 DEG C hereinafter, being carefully added into 25% ammonium hydroxide (25mL), stir 0.5 hour.Then activated carbon (1g) is added into system, mistake after 1 hour is sufficiently stirred Filter, filtrate methylene chloride extract 2 times (2 × 100mL).Merge organic phase, anhydrous sodium sulfate dries, filters, organic phase high vacuum Removed under reduced pressure solvent obtains Marbofloxacin crude product (5.4g).H is added in the crude product2In O (50mL), first acid for adjusting pH value is slowly added dropwise To 3.2 (pH meter detections), 4 hours are stood, filtering, filtrate added drop-wise sodium bicarbonate aqueous solution adjusting pH value to 6.2 (pH meter detections), A large amount of solids are precipitated, and ice salt bath cooling system stirs 1 hour to 0 DEG C or so, filtering, obtain Marbofloxacin after product drying (4.72g)。
Patent
Publication numberPriority datePublication dateAssigneeTitleUS4801584A *1986-09-121989-01-31Hoffmann-La Roche Inc.Pyrido(3,2,1-IJ)-1,3,4 benzoxadiazine derivativesCN1116849A *1993-01-231996-02-14辉瑞大药厂Process for the manufacture of a tricyclic compoundCN102060860A *2011-01-072011-05-18安徽美诺华药物化学有限公司Preparation method of MarbofloxacinCN102617595A *2012-03-232012-08-01江西华士药业有限公司Preparation method of fluoroquinolone antibacterial medicament marbofloxacinCN102712598A *2009-11-192012-10-03新梅斯托克尔卡·托瓦纳·兹德拉维尔公司A process for a preparation of marbofloxacin and intermediate thereof CN110283186A *2019-07-192019-09-27海门慧聚药业有限公司A kind of crystal form of Marbofloxacin and preparation method thereof
PATENT
CN 107522718
PATENT
CN 102617595,
PATENT
Indian Pat. Appl., 2009CH00164,
Example 2: Preparation of ethyl 6,8-difluoro-1-(N~methylfomnamido)-7-(4-methyl-1- piperazinyl)-4-oxo-4H-quinoline-3-carboxylate hydrochloride of Formula (Ilia)
STR IIIA
Water (400 ml) and the compound of Formula (IIa) (200 g) are charged into a round bottom flask at 28°C and concentrated HCI (124 ml) is added slowly at a temperature below 40°C, and the mass is heated to 95-1OO0C. 300 ml of water and ethanol are distilled under vacuum at 1004C. The mass is cooled to 25-30°C. Acetone (400 ml) Is added and the mass is cooled to 0-5°C. The mass is maintained at 0-58C for 30-60 minutes and the product is filtered. The product is washed with pre-chilled acetone (200 ml) and dried under vacuum at 70-75°C for 12-15 hours to obtain the title compound. Yield: 181.0 g (95%). Example 3: Preparation of marbofloxacin from the compound of Formula (Ilia) Ethylene glycol (100 ml) and potassium hydroxide (17.3 g) are stirred for 10- 15 minutes for dissolution. A compound of Formula (Ilia) (10 g) is added and the mass is heated to 120-130’C, and then maintained for 24 hours. The mass is cooled to 30°C and water (15 ml) is added. Hydrochloric acid (36%, 18 ml) is slowly added below 404C.rformic acid (6 ml) is slowly added below 40°C and the mass is stirred for 20-30 minutes. Formaldehyde (5 ml) is added and the mass is then heated to 70-75°C and maintained for 1-2 hours. The mass is slowly cooled to 15-20°C and stirred for 30-60 minutes. The obtained solid dihydroformate salt is filtered and the wet cake is washed with pre-chilled demineralized water (5 ml). The material is suction dried for 2-3 hours. Methanol (50 ml), demineralized water (15 ml), and the wet cake are charged into a round-bottom flask and stirred for 10-15 minutes.
Ammonia solution (25%, 7.5 ml) is added and stirred for 30-60 minutes at 25-35°C. The turbid solution is filtered and the wet cake is washed with methanol (5 ml) at 25- 35°C. The water and methanol are distilled at 60-70°C under vacuum until 20 ml remain. The mass is cooled to 0-5°C and maintained for 30-60 minutes. The solid is filtered at 0-5°C and the wet cake is washed with methanol (10 ml). The material is suction dried for 30-60 minutes and the product is dried at 60-70°C under vacuum for 18-20 hours. Yield: 6.51 g (70%). Example 4: preparation of marbofloxacin from a compound of Formula (Ilia) Ethylene glycol (150 ml) and potassium hydroxide (72.2 g) are stirred for 10- 15 minutes for dissolution. A compound of Formula (Ilia) (50 g) is added and the mass is heated to 115-1256C, and then is maintained for 10-12 hours at 115— 125°C. The mass is cooled to 25-35°C and water (150 ml) is added. Formic acid (98%, 100 m!) is slowly added below 45°C and the mass is stirred for 30-60 minutes. Formaldehyde (37-41%, 35 ml) is added to the mass, which is then heated to 70- 75°C and maintained for 1-2 hours. The mass is slowly cooled to 0-5°C and stirred for 1-2 hours. The obtained solid dihydroformate salt is filtered and the wet cake is washed with pre-chilled water (50 ml). The material is suction dried for 1 hour and washed with pre-chilled acetone (50 ml) and suction dried for 1 hour. Methanol (250 ml), water (100 ml), and the wet cake are charged into a round-bottom flask and stirred for 10-15 minutes. Ammonia solution (25%, 40 mi) is added and stirred for 30-60 minutes at 25-35°C. The turbid solution is filtered and the wet cake is washed with methanol (50 ml) at 25-35°C. The filtrate is distilled at 60-70°C under vacuum until 75-100 ml remain. The mass is cooled to 10-15’C and maintained for 30-60 minutes. The solid free base is filtered at 10-15°C and the wet cake is washed with chilled methanol (50 ml). The material is suction dried for 30-^60 minutes and the product is dried at 60-70°C under vacuum for 10-12 hours. Yield: 33.0 g (70.8%). Example 5: Preparation of marbofloxacin from a compound of Formula (Ilia) Water (350 ml) and potassium hydroxide (86.6 g) are stirred for 10 minutes. A compound of Formula (Ilia) (50 g) is added and the mass is heated to 100-104°C. The mass is maintained for 105-110 hours at 100-1040C, then is copied to 25-35°C and water (65 ml) is added. Hydrochloric acid (36%, 125 ml) is slowly added below 40°C and the mass is stirred for 30 minutes. Formaldehyde (37%, 19 ml) is added and the mass is heated to 70-756C. The mass is maintained for 1-2 hours at 70-75 0C and then is slowly cooled to 0-5°C and maintained for 30-60 minutes. The obtained solid hydrochloride salt is filtered and the bed is washed with pre-chilled water (25 ml) at 0-5°C. The material is suction dried. Ethanol (250 ml), water (75 ml), ammonia solution (25%, 38 ml) and the wet cake are charged into a round-bottom flask and stirred for 1-2 hours at 25-35° C. The turbid solution is filtered and the bed is washed with ethanol (50 ml). The filtrate is distilled at 65-70°C under vacuum until 100 ml remain. The mass is cooled to 0-5°C and maintained for 30-60 minutes. The solid free base is filtered and the wet cake is washed with pre-chilled ethanol (50 ml). The product is dried under vacuum at 60-70°C for 15-^20 hours. Yield: 23.3 g (50%).
Example 6: Preparation of marbofloxacin from a compound of Formula (IIa) Ethylene glycol (60 ml) and potassium hydroxide (28.05 g) are stirred for 10- 15 minutes for dissolution. A compound of Formula (IId) (20 g) is added. The mass is heated to 120-135°C and maintained for 4-6 hours. The mass is cooled to 30°C and water (60 ml) is added. Formic acid (98-100%, 40 ml) is slowly added below 40°C and stirred for 20-30 minutes. Formaldehyde (37-41%, 12 ml) is added to the mass, which is heated to 70-75°C and maintained for 1-2 hours. The mass is slowly cooled to O-S6C and stirred for 30-60 minutes. The obtained solid dihydroformate salt is filtered and the wet cake is washed with pre-chilled water (20 ml). The material is suction dried for 2-3 hours. Methanol (100 ml), water (30 ml), and the wet cake are charged into a round-bottom flask and stirred for 10-15 minutes. Ammonia solution (25%, 20 ml) is added and stirred for 30-60 minutes at 25-35°C. The turbid solution is filtered and the wet cake is washed with methanol (10 ml) at 25-35°C. The water and methanol are distilled at 60-70°C under vacuum until 40 ml remain. The mass is cooled to 0-5°C and maintained for 30-60 minutes. The solid free base is filtered at 0-5°C and the wet cake is washed with methanol (20 ml). The material is suction dried for 30-60 minutes and the product is dried at 60-70°C under vacuum for 18-20 hours. Yield: 12.6 g (71%)
Example 7: Purification of marbofloxacin To crude marbofloxacin (25 g) is added methanol (125 ml) and ammonia (18.75 ml). Half of the volume of the methanol and ammonia solution is removed by azeotropic distillation. The mass is slowly cooled and maintained for 1 hour. The product is filtered and washed with chilled methanol (25 ml). The product is suction dried for 30 minutes and dried under vacuum for 12 hours, to yield pure marbofloxacin of a purity 99.80%. XRD pattern, DSC thermogram, TGA1 and IR are substantially in accordance with Figs. 1, 2, 3, and 4, respectively. Yield: 22 g (88.0%),
Marbofloxacin is the common name for 9-fluoro-2,3-dihydro-3-methyl-10-(4-methyl-1-piperazinyl)-7-oxo-7H-pyridol(3,2,1-ij)(4,2,1)benzoxadiazin-6-carboxylic acid, of the formula :
[0003] Marbofloxacin is a potent antibiotic of the fluoroquinolone group.
[0004] EP 259804 describes marbofloxacin as well as a synthesis for the preparation thereof by a multistep process which is unpractical for a large scale manufacture, since it requires high temperatures and reagents not suitable for large-scale production, resulting in low over-all yields. The process for the preparation is disclosed in the reaction scheme 1.
[0005] EP 680482 discloses an alternative approach for the preparation of marbofloxacin, wherein hydroxy group is introduced into molecule by means of reaction of intermediate with alkali metal hydroxide in aqueous media. The starting material used is 2,3,4,5-tetrafluorobenzoic acid. Disadvantages of this process are relatively high excess of alkali metal hydroxide and lengthy procedure. The process for the synthesis according to this patent is shown in the reaction scheme 2.
[0006] Research Disclosure No. 291, 1988, pages 548-551 discloses an alternative route of synthesis also starting from 2,3,4,5-tetrafluorobenzoic acid. Later steps of the process are shown in the reaction scheme 3.
[0007] IT 1313683 relates to a process for preparation of marbofloxacin by a process via benzyl ether. Ether was debenzylated in aqueous solution by hydrogenating over 5% Pd/BaSO4 and the obtained product is cyclized using HCOOH/HCOH.
[0008] In view of the prior art there still exists a need for an improved method for preparation of marbofloxacin and intermediates thereof suitable for a large-scale production.
Examples
[0068] A high resolution HPLC method is used to determine an amount and purity compounds of formula I, II and IV. The tests are carried out in X-Bridge C18, 150 x 4.6mm, 3.5µm column. The mobile phase is gradient of A) 5mM NH4COOCH3 pH=7.0 B) acetonitrile. Gradient: 0’=10%B, 10’=20%B, 25′-30’=90%B, 32’=10%B.
[0069] The chromatograph is equipped with a UV detector set at 250 nm and 315nm, the flow rate is 1.0 ml per minute at 30°C.
Example 1a) 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid and 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid sodium salt
[0070]
[0071] 4.137g of Ethyl 6,8-difluoro-1-(N-methylformamido)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (10.14mmol) was put into 40mL of 10% H2SO4 and stirred at 100°C for 7 hours. Reaction mixture was cooled and crystals were formed. Mixture was cooled to 4°C and filtered with suction. Filter cake was washed with a mixture of H2O/EtOH/THF (1/1/5) and dried. 3.260g of 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid as yellow crystals were obtained (91%).
[0072] In case the sodium salt is desired the product obtained in previous step was put into 5mL of EtOH and 10mL of CH2Cl2 and 1.20g of NaOH dissolved in 2mL of water was added. Solution was stirred at room temperature. for 1h, dried with Na2SO4 and evaporated. 2.90g of pure title product was isolated (yellow powder, 7.71mmol, 76%).
b) 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
[0073]
[0074] 400mg of Ethyl 6,8-difluoro-1-(N-methylformamido)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylate (0, 979mmol) was put into 2mL of 10% H2SO4 and stirred at 100°C for 2 hours. Reaction mixture was cooled and crystals were formed. To this mixture 1,7mL of 25% aq. NH3 was slowly added. At first very dense suspension was formed that dissolves with further addition of ammonia solution. At the end clear solution formed with pH of 9. Ammonium sulphate was precipitated by the addition of 10mL of EtOH , filtered off and washed with 5mL of H2O/EtOH (1/2). Mother liquor was dried on the rotary evaporator and 10 mL of EtOH/H2O mixture (7/3) was added to precipitate residual inorganic salt, which was again filtered off. Remaining yellow solution was dried on a rotary evaporator to obtain 321mg of yellow powder (0.912 mmol, 93%).
Example 26-fluoro-8-hydroxy-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid
[0075]
[0076] 178 mg of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid sodium salt (0.470mmol) was mixed with 360 mg of Me4NOH.5H2O (2.00 mmol) and stirred at 100°C for 4 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 0.10mL of HCOOH was added to neutralize hydroxide. 5mL of EtOH is added to precipitate the product, which was filtered with suction and filter cake was washed with 2mL of cold EtOH. 90mg of the product was obtained.
Example 39-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid formate salt
[0077]
[0078] 180 mg of 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid sodium salt (0.481mmol) was mixed with 360 mg of Me4NOH.5H2O (2.00 mmol) and stirred at 100°C for 3 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 1 mL of HCOOH was added followed by addition of 0.4 mL of 37% aq. solution of HCHO and stirred at 70°C for additional hour. Reaction mixture was cooled to room temperature and 5mL of EtOH was added to precipitate the product, which was filtered with suction and filter cake was washed with 2mL of cold EtOH. 111 mg of grey powder was obtained.
Example 49-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid formate salt
[0079]
[0080] 1.14g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.00mmol) was mixed with 3.06g of Me4NOH.5H2O (16.96mmol) and stirred at 100°C for 5 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 1.44 mL of HCOOH (85% aq. sol) was added followed by addition of 0.5 mL of 37o aq. solution of HCHO and the flask was cooled on the water bath at 22°C. Another 1.44mL of 85% HCOOH was added and the reaction mixture was warmed to 70°C for 30min and after cooling 20mL of EtOH was added to the reaction mixture and left in a refrigerator for 16h. Precipitate was filtered under reduced pressure and washed with cold ethanol (10mL). After drying 1.23g of grayish powder was obtained (90%) .
Example 59-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
[0081]
[0082] 1.145g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.01mmol) was mixed with 2.72g of Me4NOH.5H2O (15.00mmol) and stirred at 100°C for 8 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved. After stirring at 70°C for 30min (precipitate was formed again after 5min) reaction flask was cooled to room temperature and 20mL of EtOH was added to the reaction mixture and left in a refrigerator for 16h. Precipitate was filtered under reduced pressure and washed with cold ethanol (10mL). After drying 1.165g of grayish powder was obtained (85%), with a purity of 97.11% (HPLC).
[0083] Crude reaction product was mixed with 0.9mL of 25% NH3 aqueous solution and crystallized in a mixture of 26mL of EtOH and 14mL H2O. 0.673g of powder was obtained (61%) with a purity of 98.75% (HPLC).
Example 69-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
[0084]
[0085] 1.140g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.00mmol) was mixed with 2.72g of Me4NOH.5H2O (15.01mmol) and stirred at 100°C for 8 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and 20mL of H2O was added to the reaction mixture and left in a refrigerator for 16h. Precipitate was filtered under reduced pressure and washed with cold ethanol (10mL). After drying 1.022g of greyish powder was obtained (75%). with a purity of 97.11% (HPLC).
[0086] Crude reaction product was mixed with 0.9mL of 25% NH3 aqueous solution and crystallised in a mixture of 20mL of EtOH and 6mL CHCl3. 0.771g of yellow powder was obtained (71%) with a purity of 99.50% as determined by HPLC.
Example 79-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
[0087]
[0088] 1.142 g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.01mmol) was mixed with 3.26g of Me4NOH.5H2O (18.01mmol) and stirred at 100°C for 4 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and dried on the rotary evaporator. 20mL of H2O was added to the reaction mixture and cooled in a refrigerator. Precipitate was filtered under reduced pressure. After drying 1.147g of white powder was obtained (84%).
[0089] Crude reaction product was mixed with 5mL of water and 2mL of 25% aqueous solution of NH3 and clear solution was obtained. To this solution, 7mL of EtOH was added and dried under reduced pressure. Product was crystallized in a mixture of 15mL of EtOH and 10mL CHCl3 to obtain 0.4321g of white powder (41%) with a purity of 98.63% as determined by HPLC
Example 89-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
[0090]
[0091] 1.136 g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (2.98mmol) was mixed with 2.73g of Me4NOH.5H2O (15.00mmol) and stirred at 100°C for 7 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and dried on the rotary evaporator. 20mL of H2O was added to the reaction mixture and cooled in a refrigerator. Precipitate was filtered under reduced pressure. After drying 1.039g of grey powder was obtained (77%).
[0092] Crude reaction product was neutralized with 2mL of 25% aqueous solution of NH3 and clear solution was diluted with 15mL of EtOH and 9mL of H2O. Solution was partially dried under reduced pressure until the formation of precipitate. At this point mixture was cooled in a refrigerator and precipitate was isolated by filtration under reduced pressure to obtain 0.675g of powder (65%) with a purity of 98.84% as determined by HPLC.
Example 99-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
[0093]
[0094] 1.140g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (3.01mmol) was mixed with 3.30g of Me4NOH.5H2O (18.20mmol) and stirred at 100°C for 4 hours. Ammonium salt melts and dark brown oil is formed during the reaction. Reaction mixture was cooled to room temperature and 3.0 mL of HCOOH was added followed by addition of 0.5 mL of 37% aq. solution of HCHO (6.0mmol) and the flask was cooled on the water bath at 22°C. Precipitate was immediately formed. The flask was warmed to 70°C, during which precipitate was dissolved and stirred for 30 min (after stirring for at 70°C for 5min precipitate formed again). Reaction flask was cooled to room temperature and dried on the rotary evaporator. 20mL of H2O was added to the reaction mixture and cooled in a refrigerator. Precipitate was filtered under reduced pressure to obtain 0.847g of solid, while mother liquid was diluted with EtOH and concentrated under reduced pressure until precipitate forms, which was filtered again to obtain additional 0.208g of solid. The yield of combined solid material is 1.055g, 77%. Crude reaction product (formate salt) was crystallized in H2O/EtOH (25mL/10mL) to obtain 0.722g (53%) of yellow powder. Formate salt was put in 20mL of EtOH/CH2Cl2 mixture (1/1) and 0.5mL of 25%aq. NH3 was added to obtain clear solution. Solution was dried with Na2SO4 and solvent evaporated under reduced pressure to obtain 0.580g of yellow powder (53%).
Example 109-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3,7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid
[0095]
[0096] 100 mL reactor with a rotary stirrer was charged with 10,16g of 6,8-difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (28,83mmol) and 26,50g of Me4NOH˙5H2O (146,25mmol) that was previously mixed together. Temperature of the heating jacket was set to 100°C and stirring to 100s-1, while water was allowed to evaporate out of the reactor during the reaction. Reaction was stirred at specified temperature for 5 hours and homogenous dark brown oil was obtained. Temperature of reactor was cooled to 20°C, 30mL of HCOOH was added and stirred well so that all oil is transformed into brown suspension. 4,5mL of 37% aq. HCHO was added drop-wise and heated at 70°C for 30min. Reaction mixture was cooled to 20°C and 20mL of water added to precipitate the product in the form of formate complex. Suspension was cooled to 0°C and filtered under reduced pressure and washed the filter cake with additional 10mL of cold water to obtain 8,38g of white powder. Mother liquor was partially evaporated under reduced pressure and when solid started to precipitate it was filtered again to obtain additional 0.80g of powder. 50mL of EtOH was added into the mother liquor to precipitate the product and after filtration at reduced pressure further 0.80g of white powder was obtained. Product was collected and 9,98g of white powder was suspended in a mixture of 50mL of EtOH and 50mL of CH2Cl2. Into the suspension 25% aq. NH3 was added to neutralize the formate complex and after addition of 12mL of NH3 all product was dissolved and small amount of solid material is formed. 5g of anhydrous Na2SO4 was added to dry the organic solution and it was filtered off and solvent evaporated under reduced pressure. 8.99g of slightly yellow powder was obtained in 86% yield.
Example 11Crystallization from ethanol/toluene/water 2:1:1
[0097] 8.4g of crude marbofloxacin was suspended in a mixture of 83 ml of ethanol, 41ml of toluene and 41 ml of water and heated to reflux. From the clear yellow solution formed 83 ml of solvent mixture was distilled off, whereby the temperature rose from 74 to about 79°C, and a yellow precipitate was formed. The suspension was cooled to 20° – 25°C, stirred for 1 hour, filtered, and the filter cake was washed with 3 portions of 6 ml of ethanol to yield after drying in vacuum dryer the product in more than 95% yield.
Example 12Crystallization of marbofloxacin starting from marbofloxacin formate
[0098] 26g of marbofloxacin formate was suspended in a mixture of 65ml of ethanol and 27ml of water. Under stirring a solution of 25% ammonia in ethanol (20ml 25%NH3/10ml EtOH) is slowly (about 30 minutes) added by drops until the substance is dissolved and pH value of 7-9 is reached. The reaction was stirred for about 15 minutes and filtered. The filtrate was evaporated at 110°C until about 60ml of the solvent was distilled off and marbofloxacin started to precipitate. After distillation the suspension was cooled and stirred for 0.5 to 1 hour at 0-5°C, filtered, to yield after drying at 40°C/50mbar for 3 to 5 hours the product in 100%yield.
Example 13Crystallization from ethanol
[0099] 1g of marbofloxacin was dissolved under heating to reflux in 160ml of ethanol, after filtration, the solution is cooled and the crystallized product is recovered in more than 90% yield.
Example 146,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid
[0100]
[0101] 10mmol of 6,7,8-Trifluoro-1-(N-methylformamido)-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester was put in the round bottomed flask. 20mL of 10% H2SO4 was added and stirred with the temperature of the sand bath of 100°C for the time periods specified in the following table. Reaction mixture was cooled down to 4°C, filtered and the cake washed with water and the conversion an yield were determined.
[0102] The experiment was repeated but starting compound was mixed with 1.0mL of solvent (EtOH, AcOH or MeCN as specified in the following table) before adding the 10% H2SO4.
[0103] The starting compound is insoluble in aqueous phase. By mixing the starting compound with a small amount of polar solvent (EtOH, MeCN, AcOH) a film is formed around the crystals which improves wetting of the crystals with the aqueous acid. Without addition of polar solvent prior to adding the aqueous acid solution wetting of the crystals is impaired and the reaction is slower.Exp.Reaction time (solvent)Conversion (yield)14.016h65%14.027h60%14.0324h100%14.0424h100% (94%)14.056h (0.1mL AcOH per mmol)91%14.066h (0.1mL EtOH per mmol)89%14.0721h (0.1mL MeCN per mmol)100% (97%)14.0821h (0.1mL MeCN per mmol)100% (96%)
Example 156,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid
[0104] 3.30g of 6,7,8-Trifluoro-1-(N-methylformamido)-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester (10.054 mmol) was put into the round-bottomed flask equipped with the magnetic stirrer. 1mL of MeCN was added and stirred for a minute. 20mL of 10% H2SO4 was added and stirred. The flask was put into the sand bath (T = 100°C) and stirred for 21h. Suspension was cooled down to 4°C and filtered under suction. Yellow powder was washed twice with cold water and dried. 2.646g of yellow powder was obtained (9.721 mmol, 96.7%) and identified by NMR spectroscopy to be title compound.
Example 166,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid
[0105] 6,7,8-Trifluoro-1-(N-methylformamido)-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid ethyl ester (6.868g, 20.92 mmol) was mixed with 1mL of EtOH (to decrease the hydrophobicity of the substrate). Next, 40mL of 10% aqueous H2SO4 solution was added and the mixture was stirred at the temperature of the bath of 100°C for 12h. A white suspension formed which was cooled to 0°C and filtered under reduced pressure. The white powder was washed with cold water and cold EtOH and dried. 5.135g of yellow powder was obtained and identified as title compound by 19F and 1H NMR spectroscopy. The yield of hydrolysis was 90%.
Example 176,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid
[0106]
[0107] 6,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid (272mg, 1.0mmol, obtained as described in Example 16, and 400 mg of N-methylpiperazine (4.0mmol) were mixed with 1mL of EtOH and stirred under reflux temperature (jacket temperature Tj=100°C). After two hours of reaction clear solution formed, afterwards the product precipitated and a very dense suspension was formed. Reaction was stopped after three hours of stirring at Tj=100°C. A sample was put directly to the NMR analysis and only two signals were observed indicating reaction was quantitative. Crude reaction product was diluted with EtOH and neutralized by addition of aqueous solution of NH3 until pH of 8 was reached. Suspension was cooled to 0°C and product isolated by filtration under reduced pressure, washed further with 10mL of cool EtOH and dried. 138mg (39%) of product was obtained.
Example 186,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid
[0108] 6,7,8-Trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid (1.087g, 3.993mmol), 484mg of N-methylpiperazine (4.83mmol) and 484 mg of Et3N (4.78mmol) were mixed with 8mL of EtOH and stirred under reflux temperature (Tj=100°C). After 19h of reflux yellow solution and white precipitate are formed in the reaction flask. Solvent was evaporated under reduced pressure and put directly to the NMR analysis. Crude reaction product was mixed with 20mL of EtOH and suspension cooled in the refrigerator. The product (white precipitate) was isolated by filtration under reduced pressure, washed further with 10mL of cool EtOH and dried. 1.178g of white powder was obtained (3.375 mmol, 800).
Example 196,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid
[0109] In accordance with examples 17 and 18 additional experiments were carried out using different reaction conditions for the conversion of 6,7,8-trifluoro-1-methylamino-1,4-dihydro-4-oxo-3-quinoline-carboxylic acid into 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid. The experiments were performed according to the following general procedure: 1.0mm of starting compound was put in the round bottomed flask and N-methylpiperazine (NMP), base and solvent were added according to the following table. Reaction mixture was stirred at the corresponding temperature. Solvent was evaporated and crude reaction mixture analyzed directly by NMR (1H and 19F).
Example 206,8-Difluoro-1-(N-methylformamido)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid ethyl ester
[0111]
[0112] Substitution:6,7,8-Trifluoro-1-(N-methylformamido)-4-oxo-1,4-quinoline-3-carboxylic acid ethyl ester (1.0 mmol, 324mg) was mixed with 2 equivalents of N-methylpiperazine (220mg) and 400mg Et3N stirred for three hours at 100°C. Reaction mixture liquefied in 10 minutes and solidified again within 30 minutes of the reaction (that is the reason for higher amount of TEA). After 3 hours of stirring was reaction mixture cooled to room temperature and analyzed by NMR spectroscopy.
[0113] Substitution: The above reaction was repeated but Et3N was replaced by 1 equivalent of DABCO.
[0114] In both cases, substitution was quantitative and analysis of the crude reaction mixtures showed that there was some hydrolysis of the ethyl ester (EE) to the free carboxylic acid (CA) group resulting in a product mixture. The results are summarized in the following table. Ethyl ester is readily soluble in water.Exp.Reaction conditionsConversion (yield)20.012.5 NMP, 1 DABCO, 100°C, 3h100% (48% EE, 52% CA)20.022.5 NMP, 4 Et3N, 100°C, 3h100% (58% EE, 42% CA)
Example 216,8-Difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid (one-pot reaction)
[0115]
[0116] 6,7,8-Trifluoro-1-(N-methylformamido)-4-oxo-1,4-quinoline-3-carboxylic acid ethyl ester (1.0 mmol, 324mg) was mixed with 2 equivalents of N-methylpiperazine (200mg) and stirred for one hour at 100°C. Reaction mixture liquefied in 10 minutes and solidified again within 30 minutes of reaction. After one hour of reaction the reaction mixture was cooled to room temperature and 10% aqueous H2SO4 (5mL) was added and stirred again at 100°C for two hours. Yellow solution was cooled to 0°C so that product precipitated. It was isolated by filtration under reduced pressure. Pure 6,8-difluoro-1-(methylamino)-7-(4-methylpiperazin-1-yl)-4-oxo-1,4-quinoline-3-carboxylic acid in the form of sulfate salt was obtained (as determined by NMR) as slightly yellow powder (279mg, 58%).
Example 22Synthesis of 9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-3, 7-dihydro-2H-[1,3,4]oxadiazino[6,5,4-ij]quinoline-6-carboxylic acid (Marbofloxacin, MBX)
[0117] 13.5 g of 6,8-Difluoro-1-(methylamino)-7-(4-methyl-piperazin-1-yl)-4-oxo-1,4-dihydroquinoline-3-carboxylic acid hydrochloride and ca. 63 g of tetramethylammonium hydroxide water solution 25 % were charged into a reactor and slowly heated to 100°C. When this temperature was reached, water was removed by distillation at reduced pressure (between 0.8 to 0.3 bar) in such a manner that ca. 25 to 32 ml of water were removed in 3 hours. The reaction mixture was stirred for another 3 hours and after completion of the conversion, the reaction mixture was cooled to 0 – 10 °C and ca. 40.5 ml of formic acid were slowly added with violent agitation. The temperature was maintained below 20°C, preferable between 0 – 10°C. Then ca. 6.1 ml of formaldehyde were slowly added. After addition the reaction mixture was heated to 70°C and maintained at this temperature for about 30 minutes.
[0118] The reaction mixture was cooled to room temperature (20 – 30°C), ca. 27 ml of purified water were added and the mixture was stirred for 30 minutes. Then the reaction mixture was cooled to 0 – 5°C and stirred at this temperature for at least 2 hours. The product marbofloxacin formate (MBXBZ) was centrifuged and washed with 10 – 15 g of cooled (0 – 5°C) purified water. The product was spun dried and collected.
[0119] Wet product MBXBZ was added to the mixture of 67 ml of ethanol, 67 ml of methylene chloride and 16.2 ml of ammonia solution (ca. 25 0). If phases did not separate, additional 63 ml of methylene chloride and 33 ml of purified water were added. The pH of the water phase was adjusted to be between 7 and 9.5, preferable between 7.5 and 8.5. The mixture was agitated for approximately 15 minutes to 1 hour and then the layers were separated and both phases were subjected to in process control (IPC) analysis.
[0120] If IPC results showed that extraction was not complete, ca. 63 ml of methylene chloride were added to the water layer and the extraction was repeated until the IPC specification was met.
[0121] The organic phases were combined and ca. 6.8 mg of sodium sulphate anhydrous and optionally 0.4 mg of activated charcoal were added. The mixture was mixed for at least 30 minutes and filtered, then organic solvent was distilled off to obtain crude marbofloxacin.
Purification of the crude Marbofloxacin
[0122] In an inert atmosphere 5 g of purified water, 12 g of ethanol 96 % and 4.3 g of toluene (ratio between the solvents was within the following ranges: ethanol : toluene : water : 1.8 – 2.8 : 1 : 1.1 – 1.2) were charged into a reactor and wet crude marbofloxacin (MBXCA) from the previous step was added under nitrogen. The mixture was slowly heated to reflux (70 – 80°C) until a clear solution was obtained. The solution was stirred for 0.5 hour under this temperature and then one half of the azeotrope solvent mixture (toluene : water : ethanol = 51 % : 6 % : 43 %) was evaporated. Then the remaining mixture was cooled slowly to 5°C (allowed interval is between 0 and 25 °C) with agitation (optionally 1 % mass of product of disodium-EDTA can be added). The mixture was mixed for 1 to 3 hours and the product was then isolated by centrifugation, washed with 13 g of ethanol, spun dry and collected. The product was dried at temperature 40 – 45°C, p < 100 mbar for 8 hour.
Example 23Purification of Marbofloxacin
[0123] Marbofloxacin was dissolved in 20 parts by weight of water by addition of acetic acid. Marbofloxacin was completely dissolved at pH of 5.3. Active charcoal was added and the mixture was stirred overnight. The mixture was then filtered using activated charcoal filter. The pH of the filtrate was adjusted to 7.2 by use of KOH, the obtained suspension was stirred for 1 hour at room temperature and then the precipitated product was recovered. Marbofloxacin with a purity of 99,9% (HPLC area) was obtained.
[0124] HPLC analysis was performed on a pentafluorophenyl propyl (PFP) column (type Luna® PFP, 150 x 4.6mm, 3µm, Phenomenex, USA); detector: UV315 nm; flow rate: 0.8 ml/min; injection volume: 5 µl; mobile phase: A: 0.02M NaH2PO4xH2O+0,1% TEA, pH2.5; B: acetonitrile : methanol = 5:95 (v/v) ; gradient: 0’=10B, 25’=100B, 30’= 100B, 32’=10B. The HPLC chromatogram of marbofloxacin prior to purification is shown in Figure 1, the HPLC chromatogram after purification is shown in Figure 2. As evident from the chromatograms all products with retention time above 24min were successfully eliminated.
Mechanism of action
Its mechanism of action is not thoroughly understood, but it is believed to be similar to the other fluoroquinolones by impairing the bacterial DNA gyrase which results in rapid bactericidal activity.[1] The other proposed mechanisms include that it acts against nondividing bacteria and does not require protein and RNA synthesis, which block protein and RNA synthesis respectively.[2]
Activity
Marbofloxacin is a synthetic, broad spectrum bactericidal agent. The bactericidal activity of marbofloxacin is concentration dependent, with susceptible bacteria cell death occurring within 20–30 minutes of exposure. Like other fluoroquinolones, marbofloxacin has demonstrated a significant post-antibiotic effect for both gram– and + bacteria and is active in both stationary and growth phases of bacterial replication.[3]
Marbofloxacin can be used both orally and topically. It is particularly used for infections of the skin, respiratory system and mammary glands in dogs and cats, as well as with urinary tract infections. For dogs, a dose ranges from 2.75 – 5.5 mg/kg once a day. The duration of treatment is usually at least five days, longer if there is a concurrent fungal or yeast infection.[4] Maximum duration of treatment is 30 days.[3]
Contraindications and side effects
Marbofloxacin should usually be avoided in young animals because of potential cartilage abnormalities. In rare occasion, it can cause central nervous system (CNS) stimulation and should be used with caution in patients with seizure disorders.[3] Under certain conditions it can cause discomfort such as cramps, treatable with diazepam. Other adverse effects are usually limited to gastrointestinal tract (GI) distress (vomiting, anorexia, soft stools, diarrhoea) and decreased activity.[3]
References
^ Boothe, D.M. (2001) Antimicrobial drugs. In Small Animal ClinicalPharmacology and Therapeutics, pp. 150–173. W. B. Saunders Co., Philadelphia, PA.
^ Hunter RP, Koch DE, Coke RL, Carpenter JW, Isaza R. Identification and comparison of marbofloxacin metabolites from the plasma of ball pythons (Python regius) and blue and gold macaws (Ara ararauna). J Vet Pharmacol Ther. 2007 Jun;30(3):257-62.
^ Jump up to:abcd Plumb DC (ed). Plumb’s Veterinary Handbook, 7th ed. Ames, IA: Wiley-Blackwell Publishing, 2011.
Aviptadil had been in phase II clinical trials for the treatment of pulmonary arterial hypertension and idiopathic pulmonary fibrosis. But these researches were discontinued in 2011.
In 2006, Orphan Drug Designations were granted in the E.U. for the treatment of pulmonary arterial hypertension, and sarcoidosis and acute lung injury in 2006, and 2008, respectively.
The compound was co-developed by Lung Rx (subsidiary of United Therapeutics) and Mondobiotech.
Studies have found that aviptadil may be beneficial for severely ill patients with COVID-19 related ARDS.[2]ACTIV-3, a trial examining aviptadil acetate (Zyesami), is recruiting patients as of 2 July 2021.[3] A separate trial is examining inhaled aviptadil for patients with high risk for ARDS, is ongoing as of 21 May 2021.[4] A trial for intravenous aviptadil for the same indication concluded in February 2021.[5]
U.S.-Israeli NeuroRx Inc partnered with Relief Therapeutics to develop aviptadil in the United States. In June 2020, the U.S. Food and Drug Administration granted fast-track designation to aviptadil for treatment of respiratory distress in COVID-19.[6] In September 2020, NeuroRX submitted a request for an Emergency Use Authorization to the US FDA for its use in patients in intensive care.[7]May 2021: NRx Pharmaceuticals Announces Positive Results for ZYESAMI (Aviptadil-acetate) and Submits Emergency Use Authorization Application to USFDA to Treat Critical COVID-19 in Patients Suffering from Respiratory Failure.[8]
Jan, 2021: Zuventus healthcare Ltd seeks approval for Aviptadil from India’s drug controller for emergency use in COVID-19 treatment. Mumbai’s Zuventus healthcare Ltd. has got the nod to conduct Phase 3 clinical trials of Aviptadil injectable formulation. The SEC noted that Zuventus had presented revised Phase 3 clinical trial protocol before the committee, and after “detailed deliberation”, it recommended grant of permission of Phase 3 trials with the drug.[9][10]
Aviptadil/phentolamine combination for Erectile Dysfunction (ED)
October 2000 UK (Invicorp): Aviptadil, an injectable formulation of vasoactive intestinal polypeptide (VIP) in combination with the adrenergic drug phentolamine is approved as an effective alternative therapy for erectile dysfunction (ED) patients. 1 dose intracavernosal injection contains 25 micrograms aviptadil and 2 mg of phentolamine mesilate for the treatment of erectile dysfunction. Aviptadil dose used for treatment of erectile dysfunction is far lesser as compared to dose used for the treatment of ARDS.[11][12]
Vasoactive intestinal peptide (VIP)
Vasoactive intestinal peptide (VIP) is a 28-residue amino acid peptide first characterized in 1970 that was initially isolated from porcine duodenum. A member of the secretin/glucagon hormone superfamily. VIP was initially discovered owing to its potent vasodilatory effects (as its name implies). VIP is widely distributed in the central and peripheral nervous system as well as in the digestive, respiratory, reproductive, and cardiovascular systems as a neurotransmitter and neuroendocrine releasing factor. These effects contribute to an extensive range of physiological and pathological processes related to development, growth, and the control of neuronal, epithelial, and endocrine cell function.[13]
VIP Receptors
VIP acts on two receptors – VPAC1 and VPAC2, which are class B of G-protein-coupled receptors (GPCRs).VPAC1 is mainly present in the lung and T-lymphocytes, whereas VPAC2 is mainly seen in the smooth muscle,mast cells and the basal parts of the lung mucosa.[14]
Expression of VIP
VIP is produced in the neurons in the central and peripheral nervous systems. VIP is mainly localized in the myenteric and submucosal neurons and nerve terminals in the GI tract. Endogenous VIP is released by numerous stimuli such as acetylcholine (ACh), ATP, serotonin (5-HT), substance P (SP), GLP-2 from at least two populations of VIP-positive nerves: cholinergic and non-cholinergic VIP-releasing nerves. In guinea pig small intestine, most VIP-positive nerves in the mucosa and submucosa are non-cholinergic secretomotor neurons and well colocalized with neuronal nitric oxide synthase (nNOS) in human colonic circular muscles. VIP is also expressed in immune cells, such as activated T cells and therefore present in lymphoid tissues including Peyer’s patches, the spleen, and lymph nodes, in addition to the VIP-ergic innervation in lymphoid tissues. Beside the neuronal source, VIP is also expressed and released from endocrine organs – Heart, Thyroid, Kidney and GI tracts.[15]
Localization of VIP
VIP is highly localised in lungs (70%) and binds with alveolar type II (AT II) cells via VPAC1.[2] The biological (vasodilator) activity of vasoactive intestinal peptide (VIP) was discovered in the lungs before the peptide was isolated and chemical identity characterized from intestine. Although VIP levels are consideralbly high in the brain or gut:VIP is localized in key sites in the lung, has potent activities on its major functions, and appears to play an important role in pulmonary physiology and disease.[16]
The principal localization of VIP-containing neurons in the tracheobronchial tree is in the smooth muscle layer, around submucosal mucous glands and in the walls of pulmonary and bronchial arteries. Immunoreactive VIP is also present in neuronal cell bodies forming microganglia that provide a source of intrinsic innervation of pulmonary structures.[16]
Vasoactive Intestinal Peptide (VIP) and SARS-CoV-2
VIP is highly localised in lungs and binds with alveolar type II (AT II) cells via VPAC1 receptor. AT II cells constitute only 5% of pulmonary epithelium. Angiotensin Converting Enzyme 2 (ACE 2) surface receptors arepresent in AT II cells. AT II cells produces surfactant and plays an important role in the maintenance of type 1epithelial cells. SARS-CoV-2 enters into AT II cells by binding to ACE 2 surface receptors with its spike protein. SARS CoV-2 attack mainly type II cells (not type I alveolar cells) and results in the death of alveolar type II (AT 11) cells which produces surfactant, resulting in[2]
Profound defect in oxygenation
Leading to hypoxia
Mechanism of action of Aviptadil
Pulmonary alveolar type II Cells have a high concentration of ACE 2 receptors on their cell membrane
Investigators have confirmed that the SARS-CoV family of viruses selectively attack pulmonary Alveolar Type II (ATII) cells because of their ACE2 receptors, in contrast to other pulmonary epithelial cells.
SARS-CoV Viruses bind to ACE2 receptors in order to enter the cell. Viral replication and rupture liberates inflammatory cytokines and destroys surfactant production
VIP binds uniquely to receptors on Alveolar Type II cells in the lung, the same cells that bind the SARS-CoV-2 virus via their ACE2 receptors
VIP is heavily concentrated in the lung and binds specifically to VIP receptors on alveolar type II cells. VIP exerts a broad anti-cytokine effect on immune system cells
VIP specifically upregulates surfactant production via upregulation of C-Fos protein and protects type II cells from cytokine
Upregulating the production of surfactant, the loss of which is increasingly implicated in COVID-19 respiratory failure [17]
Aviptadil a synthetic form of VIP results in rapid clinical recovery in patients with SARS-CoV-2 infection.[2]
Effect of Aviptadil on Lungs in COVID-19
Preservation of Pulmonary Tissue
Preserving surfactant production in the lung and in protecting type 2 alveolar cells. Significantly delayed the onset of edematous lung injury, effective in preventing ischemia-reperfusion injury, Prevents NMDA-induced caspase-3 activation in the Lung.[18]
Inhibits alveolar epithelial cell Apoptosis
VIP is a proven inhibitor of activation-induced perforin, as well as of granzyme B and therefore actively contributes to the reduction of deleterious proinflammatory and cell death-inducing processes, particularly in the lungs. Aviptadil restores barrier function at the endothelial/alveolar interface and thereby protects the lung and other organs from failure.[18]
VIP Promotes synthesis of pulmonary surfactant
Studies have demonstrated that VIP binds on type II cells and increases the incorporation of methyl-choline into phosphatidylcholine – the major component of the pulmonary surfactants by enhancing the activity of the enzyme choline-phosphate cytidylyltransferase. VIP upregulates C-Fos protein expression in cultured type II alveolar cells, which is instrumental in promoting synthesis of pulmonary surfactant phospholipids (Li 2007) and induces surfactant protein A expression in ATII cells through activation of PKC/c-Fos pathway.[18]
VIP decreases Pulmonary Inflammation
Anti-cytokine effect- Inhibits IL-6,TNF-α production and inhibit NF-kB activation. Protects against HCl-induced pulmonary edema.[18]
Pharmacokinetic Properties
Half-life: Its plasma half-life of elimination is 1 to 2 minutes.[2]Metabolism/Distribution: After injection of 1 µg radioactively labelled Aviptadil as bolus to patients a very rapid tissue distribution was observed Within 30 min about 45% of the radioactivity was found in the lungs Over an observation period of 24 hrs only minimal activity was detected in the GI tract & almost no activity was found in the liver or spleen Radioactivity in the lungs decreased within four hours to 25% and within 24 hours to 10% Apparent volume of distribution: Aviptadil has a volume of distribution of 14 ml/kg.[2]Tissue Distribution:Aviptadil binds to its receptors in discrete locations within the gastrointestinal, respiratory, and genital tracts. Aviptadil is localized on respiratory epithelium, smooth muscles of the airways, blood vessels and alveolar walls. Elimination:After injection of radiolabelled Aviptadil radioactivity was almost completely eliminated by the kidneys, 35% within 4 hours, and 90% within 24 hours
Justification for Aviptadil use in the treatment of ARDS
COVID-19-related death is primarily caused by Acute Respiratory Distress Syndrome (ARDS). The trigger for ARDS is widely attributed to a cytokine storm in the lungs, in which the virus causes release of inflammatory cytokines. As a result, alveolae of the lungs fill with fluid and become impermeable to oxygen, even in the setting of mechanical ventilation. SARS-CoV-2 is known to cause respiratory failure, which is the hallmark of Acute COVID-19. Tragically, survival of patients with COVID-19 who progress to Acute Respiratory Distress is dismal. There is an urgent need for a treatment approach that goes right into the heart of the matter – the alveolar type 2 cells which are vulnerable entry points and hosts for the SARS-CoV-2 virus.[19]
Aviptadil-Evidence from Studies in ARDS
Phase III Study-Increased Recovery and Survival in Patients With COVID-19 Respiratory Failure Following Treatment with Aviptadil
A multicenter, randomized, placebo-controlled trial in 196 patients with PCR+ COVID-19 receiving intensive care at 10 U.S. hospitals – 6 tertiary care and 4 regional hospitals to determine whether intravenous aviptadil (synthetic VIP) is superior to placebo in achieving recovery from respiratory failure and survival at 60 days post treatment. Primary, prespecified endpoint was “alive and free from respiratory failure at day 60.” Across all patients and sites of care, patients treated with aviptadil were significantly more likely to be alive and free from respiratory failure at 60 days, compared to those treated with placebo (P=.02) and demonstrated improvement in survival alone (P<.001). Advantages in survival for aviptadil-treated patients were seen in both the subgroup classified as 2 on the National Institute of Allergy and Infectious Disease (NIAID) ordinal scale (58.6% vs. 0%; p=.001) and the NIAID=3 subgroup (83.1% vs. 62.8%; p=.03). Among patients who recovered successfully, those treated with Aviptadil had a median 10-day reduction in length of hospital stay compared to placebo patients (P=.025). Treatment with aviptadil demonstrates multi-dimensional efficacy in improving the likelihood of recovery from respiratory failure and survival to 60 days, and markedly reduced hospital stay in critically ill patients with respiratory failure caused by COVID-19.[20]
Case report: Rapid Clinical Recovery from Critical COVID-19 Pneumonia with Aviptadil
A 54 year old man with double lung transplant presented with headache, fever and productive cough. COVID-19 infection was confirmed by positive RT-PCR of nasopharyngeal swab. The patient required only supportive care for 3 days and was discharged home. Two weeks later he presented with worsening dyspnea, fever and severe hypoxemia requiring high flow O2 and ICU admission. Chest CT showed diffuse bilateral consolidations. He had markedly elevated inflammatory markers. He was treated with dexamethasone and tocilizumab without improvement. He was not a candidate for Remdesivir due to chronic kidney disease. Convalescent plasma was not available, Pro-BNP level was normal; echocardiogram showed preserved biventricular function. He received Aviptadil, a total of three doses, per an open label access under an emergency use approved by USFDA. Rapid improvement in oxygenation and radiologic findings were noticed. No adverse effects were recorded. Patient was transferred out of the ICU 24 hours following the third dose and discharged home on room air 15 days later. This case report of lung transplant recipient with critical COVID-19 pneumonia treated with Aviptadil demonstrates rapid clinical and radiologic improvement.This is consistent with that VIP protects ATII cells, ameliorating the inflammation and improving oxygenation in critical COVID-19 pneumonia.[21]
Posology and method of administration
Aviptadil intravenous infusion is administered by infusion pump in escalating doses for 3 successive days
Day 1 : Aviptadil 0.166 mcg/kg/hr (equivalent to 1 vial of Aviptadil Injection)
Day 2 : Aviptadil 0.332 mcg/kg/hr (equivalent to 2 vials of Aviptadil Injection)
Day 3 : Aviptadil 0.498 mcg/kg/hr (equivalent to 3 vials of Aviptadil Injection)
Duration of infusion depends on the patient’s body weight
Body weight < 60 kg – 14 hour infusions of Aviptadil at escalating doses on 3 successive days
Body weight 60 – 90 kg – 12 hour infusions of Aviptadil at escalating doses on 3 successive days
Body weight > 90 kg – 10 hour infusions of Aviptadil at escalating doses on 3 successive days
^ Keijzers GB (April 2001). “Aviptadil (Senatek)”. Current Opinion in Investigational Drugs. 2 (4): 545–9. PMID11566015. Archived from the original on 2010-09-02. Retrieved 2020-04-01.
^ Javitt, Jonathan C (2020-07-25). “Vasoactive Intestinal Peptide treats Respiratory Failure in COVID-19 by rescuing the Alveolar Type II cell”. doi:10.22541/au.159569209.99474501. S2CID221509046.
^ Jump up to:ab Youssef, Jihad G.; Lee, Richard; Javitt, Jonathan; Lavin, Philip; Lenhardt, Rainer; Park, David J; Perez Fernandez, Javier; Morganroth, Melvin; Jayaweera, Dushyantha (2021). “Increased Recovery and Survival in Patients With COVID-19 Respiratory Failure Following Treatment with Aviptadil: Report #1 of the ZYESAMI COVID-19 Research Group”. SSRN3830051.
chloromethyl (8S,9S,10R,11S,13S,14S,17R)-17-ethoxycarbonyloxy-11-hydroxy-10,13-dimethyl-3-oxo-7,8,9,11,12,14,15,16-octahydro-6H-cyclopenta[a]phenanthrene-17-carboxylate(11b,17a)-17-[(Ethoxycarbonyl)oxy]-11-hydroxy-3-oxo-androsta-1,4-diene-17-carboxylic acid chloromethyl ester (11b,17a)-17-[(Ethoxycarbonyl)oxy]-11-hydroxy-3-oxoandrosta-1,4-diene-17-carboxylic Acid Chloromethyl Ester (8S,9S,10R,11S,13S,14S,17R)-17-[(éthoxycarbonyl)oxy]-11-hydroxy-10,13-diméthyl-3-oxo-6,7,8,9,10,11,12,13,14,15,16,17-dodécahydro-3H-cyclopenta[a]phénanthrène-17-carboxylate de chlorométhyle 129260-79-3[RN] 17a-Ethoxycarbonyloxy-D’-cortienic Acid Chloromethyl Ester 82034-46-6[RN] Androsta-1,4-diene-17-carboxylic acid, 17-((ethoxycarbonyl)oxy)-11-hydroxy-3-oxo-, chloromethyl ester, (11β,17α)- Androsta-1,4-diene-17-carboxylic acid, 17-[(ethoxycarbonyl)oxy]-11-hydroxy-3-oxo-, chloromethyl ester, (11β,17α)- Loteprednol Etabonate CAS Registry Number: 82034-46-6 CAS Name: (11b,17a)-17-[(Ethoxycarbonyl)oxy]-11-hydroxy-3-oxoandrosta-1,4-diene-17-carboxylic acid chloromethyl ester Additional Names: chloromethyl 17a-ethoxycarbonyloxy-11b-hydroxyandrosta-1,4-diene-3-one-17b-carboxylate; 17a-ethoxycarbonyloxy-D¢-cortienic acid chloromethyl ester Manufacturers’ Codes: CDDD-5604; HGP-1; P-5604 Trademarks: Alrex (Bausch & Lomb); Lotemax (Bausch & Lomb) Molecular Formula: C24H31ClO7, Molecular Weight: 466.95 Percent Composition: C 61.73%, H 6.69%, Cl 7.59%, O 23.98% Literature References: Ophthalmic corticosteroid. Prepn: N. S. Bodor, BE889563 (1981 to Otsuka); idem,US4996335 (1991). Physicochemical properties: M. Alberth et al.,J. Biopharm. Sci.2, 115 (1991). HPLC determn in plasma and urine: G. Hochhaus et al.,J. Pharm. Sci.81, 1210 (1992). NMR structural studies: S. Rachwal et al.,Steroids61, 524 (1996); idem et al., ibid. 63, 193 (1998). Metabolism and transdermal permeability: N. Bodor et al.,Pharm. Res.9, 1275 (1992). Evaluation of effect on intraocular pressure: J. D. Bartlett et al.,J. Ocul. Pharmacol.9, 157 (1993). Clinical trial in keratoconjunctivitis sicca: S. C. Pflugfelder et al.,Am. J. Ophthalmol.138, 444 (2004). Review of ophthalmic clinical studies: J. F. Howes, Pharmazie55, 178-183 (2000). Properties: Crystals from THF + hexane, mp 220.5-223.5°. Soly at 25° (mg/ml): 0.0005 in water; 0.037 in 50% propylene glycol + water. Lipophilicity (log K): 3.04. Melting point: mp 220.5-223.5° Therap-Cat: Anti-inflammatory (topical). Keywords: Glucocorticoid. Research Code:HGP-1; CDDD-5604; P-5604Trade Name:Lotemax® / Alrex®MOA:CorticosteroidIndication:Acne rosacea; Superficial punctate keratitis; Postoperative inflammation and pain following ocular surgery; Iritis; Herpes zoster keratitis; Allergic conjunctivitis; CyclitisCompany:Bausch & Lomb (Originator)Sales:ATC Code:S01BA14
Loteprednol etabonate was approved by the U.S. Food and Drug Administration (FDA) on Mar 9, 1998. It was developed and marketed as Lotemax® by Bausch & Lomb.
Loteprednol etabonate is a corticosteroid used in ophthalmology. It is indicated for the treatment of steroid responsive inflammatory conditions of the palpebral and bulbar conjunctiva, cornea and anterior segment of the globe such as allergic conjunctivitis, acne rosacea, superficial punctate keratitis, herpes zoster keratitis, iritis, cyclitis, selected infective conjunctivitides.
Lotemax® is available as drops for ophthalmic use, containing 0.5% of Loteprednol etabonate. The recommended dose is one to two drops into the conjunctival sac of the affected eyes four times daily.
Loteprednol (as the esterloteprednol etabonate) is a corticosteroid used to treat inflammations of the eye. It is marketed by Bausch and Lomb as Lotemax[1] and Loterex.
It was patented in 1980 and approved for medical use in 1998.[2]
Loteprednol Etabonate is the etabonate salt form of loteprednol, an ophthalmic analog of the corticosteroid prednisolone with anti-inflammatory activity. Loteprednol etabonate exerts its effect by interacting with specific intracellular receptors and subsequently binds to DNA to modify gene expression. This results in an induction of the synthesis of certain anti-inflammatory proteins while inhibiting the synthesis of certain inflammatory mediators. Loteprednol etabonate specifically induces phospholipase A2 inhibitory proteins (collectively called lipocortins), which inhibit the release of arachidonic acid, thereby inhibiting the biosynthesis of potent mediators of inflammation, such as prostaglandins and leukotrienes.
Loteprednol etabonate is an etabonate ester, an 11beta-hydroxy steroid, a steroid ester, an organochlorine compound, a steroid acid ester and a 3-oxo-Delta(1),Delta(4)-steroid. It has a role as an anti-inflammatory drug. It derives from a loteprednol.
Loteprednol Etabonate (LE) is a topical corticoid anti-inflammatory. It is used in ophthalmic solution for the treatment of steroid responsive inflammatory conditions of the eye such as allergic conjunctivitis, uveitis, acne rosacea, superficial punctate keratitis, herpes zoster keratitis, iritis, cyclitis, and selected infective conjunctivitides. As a nasal spray, it can be used for the treatment and management of seasonal allergic rhinitis. Most prescription LE products, however, tend to be indicated for the treatment of post-operative inflammation and pain following ocular surgery. A number of such new formulations that have been approved include Kala Pharmaceutical’s Inveltys – the first twice-daily (BID) ocular corticosteroid approved for this indication, designed specifically to enhance patient compliance and simplified dosing compared to all other similar ocular steroids that are dosed four times daily. Moreover, LE was purposefully engineered to be a ‘soft drug’, one that is designed to be active locally at the site of administration and then rapidly metabolized to inactive components after eliciting its actions at the desired location, thereby subsequently minimizing the chance for adverse effects.
Approval Date
Approval Type
Trade Name
Indication
Dosage Form
Strength
Company
Review Classification
2012-09-28
New dosage form
Lotemax
Postoperative inflammation and pain following ocular surgery
Gel
0.5%
Bausch & Lomb
2011-04-15
New dosage form
Lotemax
Postoperative inflammation and pain following ocular surgery
Allergic conjunctivitis,Acne rosacea,Superficial punctate keratitis,Herpes zoster keratitis,Iritis,Cyclitis,Postoperative inflammation and pain following ocular surgery
Suspension
滴眼剂,0.5%(2.5ml:12.5mg,5ml:25mg)
Bausch & Lomb
2011-11-05
Marketing approval
露达舒/Lotemax
Allergic conjunctivitis,Acne rosacea,Superficial punctate keratitis,Herpes zoster keratitis,Iritis,Cyclitis,Postoperative inflammation and pain following ocular surgery
The most common adverse effects in patients being treated with the gel formulation are anterior chamber inflammation (in 5% of people), eye pain (2%), and foreign body sensation (2%).[5]
Interactions
Because long term use (more than 10 days) can cause increased intraocular pressure, loteprednol may interfere with the treatment of glaucoma. Following ocular administration, the drug is very slowly absorbed into the blood, therefore the blood level is limited to an extremely small concentration, and interactions with drugs taken by mouth or through any route other than topical ophthalmic are very unlikely.[1]
Neither loteprednol etabonate nor its inactive metabolites Δ1–cortienic acid and Δ1-cortienic acid etabonate are detectable in the bloodstream, even after oral administration. A study with patients receiving loteprednol eye drops over 42 days showed no adrenal suppression, which would be a sign of the drug reaching the bloodstream to a clinically relevant extent.[1]
Loteprednol etabonate was developed using retrometabolic drug design. It is a so-called soft drug, meaning its structure was designed so that it is predictably metabolised to inactive substances. These metabolites, Δ1-cortienic acid and its etabonate, are derivatives of cortienic acid, itself an inactive metabolite of hydrocortisone.[1][4][6]
Cortisol, a naturally occurring corticosteroid, known as hydrocortisone when used as a drug
Δ1-Cortienic acid, inactive metabolite of loteprednol
Cortienic acid, inactive metabolite of hydrocortisone
Chemistry
Loteprednol etabonate is an ester of loteprednol with etabonate (ethyl carbonate). The pure chemical compound has a melting point between 220.5 °C (428.9 °F) and 223.5 °C (434.3 °F). Its solubility in water is 1:2,000,000,[4] therefore it is formulated for ophthalmic use as either an ointment, a gel, or a suspension.[7]
Loteprednol is a corticosteroid. The ketone side chain of classical corticosteroids such as hydrocortisone is replaced by a cleavable ester, which accounts for the rapid inactivation.[8] (This is not the same as the etabonate ester.)
^ Bodor N, Buchwald P (2002). “Design and development of a soft corticosteroid, loteprednol etabonate”. In Schleimer RP, O’Byrne PM, Szefler SJ, Brattsand R (eds.). Inhaled Steroids in Asthma. Optimizing Effects in the Airways. Lung Biology in Health and Disease. 163. Marcel Dekker, New York. pp. 541–564.
^ Pavesio CE, Decory HH (April 2008). “Treatment of ocular inflammatory conditions with loteprednol etabonate”. The British Journal of Ophthalmology. 92 (4): 455–9. doi:10.1136/bjo.2007.132621. PMID18245274. S2CID25873047.
^ Druzgala P, Hochhaus G, Bodor N (February 1991). “Soft drugs–10. Blanching activity and receptor binding affinity of a new type of glucocorticoid: loteprednol etabonate”. The Journal of Steroid Biochemistry and Molecular Biology. 38 (2): 149–54. doi:10.1016/0960-0760(91)90120-T. PMID2004037. S2CID27107845.
Further reading
Stewart R, Horwitz B, Howes J, Novack GD, Hart K (November 1998). “Double-masked, placebo-controlled evaluation of loteprednol etabonate 0.5% for postoperative inflammation. Loteprednol Etabonate Post-operative Inflammation Study Group 1”. Journal of Cataract and Refractive Surgery. 24 (11): 1480–9. doi:10.1016/s0886-3350(98)80170-3. PMID9818338. S2CID24423725.
The alkylation of pilosine (I) with ethyl chloride (II) by means of LDA in THF gives trans-pilocarpine (III), which is isomerized with LDA in THF, yielding a mixture of cis- and trans-pilocarpine (IV). Finally, this mixture is resolved by crystallization with di-p-toluoyl tartaric acid.
Schmidt, Theresa et alFrom Molecules, 26(12), 3676; 2021
Figure 1. Structure of natural occurring pilocarpine (+)-1 and its enantiomer (–)-1.
Scheme 1. Reactions and conditions: (a) hν, Bengal rosa, 8 h, 20 °C, 76% (of 3) and 5% (of 4); (b) CH2(OCH3)2, P4O10, DCM, 20 °C, 5 h, 98%; (c) CH2(OCH3)2, P4O10, DCM, 20 °C, 5 h, 99%; (d) THF, Na, 25 °C, 15 h, 72%; (e) CH2(OCH3)2, P4O10, DCM, 20 °C, 5 h, 77% (of 6) and 19% (of 7); (f) HBr, reflux, 2 d, 83%; (g) HBr, reflux, 4 d, 4%.
Scheme 2. Reactions and conditions: (a) SOCl2, reflux, 3 h, quant.; (b) Hex-OH, reflux, 16 h, 98%; (c) Rh/Al2O3, H2 (1 at), THF, 5 d, quant.; (d) Lipase PS, pH = 7.0, 2 d, 22 °C, 48% (of (±)-16) and 42% (of (–)-17); (e) PLE, pH = 7.0, 22 °C, 2 d, 96%; (f) N-methylmorpholine, iBu-chloroformate, N,O-dimethylhydroxylamine hydrochloride, 23 °C, 1 d, 84% (of (+)-18) and 85% of (–)-18); (g) LiAlH4, Et2O, 23 °C, 30 min, 95% (of (+)-19) and 95% of (–)-19; (h) CH3NH2, TosMic, DCM, benzene, NEt3, 7 d, 23 °C, 59% (of (+1)-1 and 60% of (–)-1; Hex stands for n-hexyl.
(+)-Pilocarpine [(+)-1]
Following the procedure given for the synthesis of its enantiomer, (+)-1 (1.92 g, 59%) was obtained as a colorless oil; Rf = 0.60 (SiO2, DCM/MeOH/aq NH4OH (25%), 95:4:1); [α]D = +115.7° (c 0.6, CHCl3), ee > 99% (by HPLC, Chiralcel OC, n-hexane/ethanol, 3:7, 0.3 mL/min, UV-detection λ = 215 nm; tR = (+)-1 47.1 min, tR = (–)-1 = 52.32 min); IR (film), 1H-NMR, 13C-NMR and MS (ESI, MeOH) were identical to the enantiomer (vide supra); analysis calcd. for C11H16N2O2 (208.26): C 63.44, H 7.74, N 13.45; found: C 63.31, H 7.98, N 13.32 PAPERBy Fuerstner, AloisFrom e-EROS Encyclopedia of Reagents for Organic Synthesis, 1-7; 2001
It may be used to treat a form of dry eye called aqueous deficient dry eye (ADDE)[18]
Surgery
Pilocarpine is sometimes used immediately before certain types of corneal grafts and cataract surgery.[19][20] In ophthalmology, pilocarpine is also used to reduce symptomatic glare at night from lights when the patient has undergone implantation of phakic intraocular lenses; the use of pilocarpine would reduce the size of the pupils, partially relieving these symptoms.[dubious – discuss] The most common concentration for this use is pilocarpine 1%.[citation needed] Pilocarpine is shown to be just as effective as apraclonidine in preventing intraocular pressure spikes after laser trabeculoplasty.[21]
Presbyopia
In 2021, the US Food and Drug Administration approved pilocarpine hydrochloride as an eyedrop treatment for presbyopia, age-related difficulty with near-in vision. Marketed as vuity, the effect lasts for 7 to 10 hours.[22]
Use of pilocarpine may result in a range of adverse effects, most of them related to its non-selective action as a muscarinic receptor agonist. Pilocarpine has been known to cause excessive salivation, sweating, bronchial mucus secretion, bronchospasm, bradycardia, vasodilation, and diarrhea. Eye drops can result in brow ache and chronic use in miosis.
Pharmacology
Pilocarpine is a drug that acts as a muscarinic receptor agonist. It acts on a subtype of muscarinic receptor (M3) found on the iris sphincter muscle, causing the muscle to contract – resulting in pupil constriction (miosis). Pilocarpine also acts on the ciliary muscle and causes it to contract. When the ciliary muscle contracts, it opens the trabecular meshwork through increased tension on the scleral spur. This action facilitates the rate that aqueous humor leaves the eye to decrease intraocular pressure. Paradoxically, when pilocarpine induces this ciliary muscle contraction (known as an accommodative spasm) it causes the eye’s lens to thicken and move forward within the eye. This movement causes the iris (which is located immediately in front of the lens) to also move forward, narrowing the Anterior chamber angle. Narrowing of the anterior chamber angle increases the risk of increased intraocular pressure.[24]
Society and culture
Preparation
Plants in the genus Pilocarpus are the only known sources of pilocarpine, and commercial production is derived entirely from the leaves of Pilocarpus microphyllus (Maranham Jaborandi). This genus grows only in South America, and Pilocarpus microphyllus is native to several states in northern Brazil.[25]
Pilocarpine is extracted from the powdered leaf material in a multi-step process. First the material is treated with ethanol acidified with hydrochloric acid, and the solvents removed under reduced pressure. The resultant aqueous residue is neutralized with ammonia and put aside until the resin has completely settled. It is then filtered and concentrated by sugar solution to a small volume, made alkaline with ammonia, and finally extracted with chloroform. The solvent is removed under reduced pressure.[verification needed]
Cost
Pilocarpine is one of the lowest cost medications for glaucoma.[26]
Trade names
Pilocarpine is available under several trade names such as: Diocarpine (Dioptic), Isopto Carpine (Alcon), Miocarpine (CIBA Vision), Ocusert Pilo-20 and -40 (Alza), Pilopine HS (Alcon), Salagen (MGI Pharma), Scheinpharm Pilocarpine (Schein Pharmaceutical), Timpilo (Merck Frosst) and Vuity (Abbvie).
Research
Pilocarpine is used to induce chronic epilepsy in rodents, commonly rats, as a means to study the disorder’s physiology and to examine different treatments.[27][28] Smaller doses may be used to induce salivation in order to collect samples of saliva, for instance, to obtain information about IgA antibodies.
Veterinary
Pilocarpine is given in moderate doses (about 2 mg) to induce emesis in cats that have ingested foreign plants, foods, or drugs. One feline trial determined it was effective, even though the usual choice of emetic is xylazine.
References
^ Jump up to:abcdefg“Pilocarpine”. The American Society of Health-System Pharmacists. Archived from the original on 28 December 2016. Retrieved 8 December 2016.
^ Gornitsky M, Shenouda G, Sultanem K, Katz H, Hier M, Black M, Velly AM (July 2004). “Double-blind randomized, placebo-controlled study of pilocarpine to salvage salivary gland function during radiotherapy of patients with head and neck cancer”. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology, and Endodontics. 98 (1): 45–52. doi:10.1016/j.tripleo.2004.04.009. PMID15243470.
^“Glaucoma and ocular hypertension. NICE guideline 81”. National Institute for Health and Care Excellence. November 2017. Retrieved 19 September 2019. Ocular hypertension… alternative options include carbonic anhydrase inhibitors such as brinzolamide or dorzolamide, a topical sympathomimetic such as apraclonidine or brimonidine tartrate, or a topical miotic such as pilocarpine, given either as monotherapy or as combination therapy.
^ Rosin A (1991). “[Pilocarpine. A miotic of choice in the treatment of glaucoma has passed 110 years of use]”. Oftalmologia (in Romanian). 35 (1): 53–5. PMID1811739.
^ Holmstedt, B; Wassén, SH; Schultes, RE (January 1979). “Jaborandi: an interdisciplinary appraisal”. Journal of Ethnopharmacology. 1 (1): 3–21. doi:10.1016/0378-8741(79)90014-x. PMID397371.
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^“Pilocarpine”. MedLinePlus. U.S. National Library of Medicine. Archived from the original on 2010-03-06.
^ Yang, WF; Liao, GQ; Hakim, SG; Ouyang, DQ; Ringash, J; Su, YX (1 March 2016). “Is Pilocarpine Effective in Preventing Radiation-Induced Xerostomia? A Systematic Review and Meta-analysis”. International Journal of Radiation Oncology, Biology, Physics. 94 (3): 503–11. doi:10.1016/j.ijrobp.2015.11.012. hdl:10722/229069. PMID26867879.
^ De Abreu IN, Sawaya AC, Eberlin MN, Mazzafera P (November–December 2005). “Production of Pilocarpine in Callus of Jaborandi (Pilocarpus microphyllus Stapf)”. In Vitro Cellular & Developmental Biology – Plant. Society for In Vitro Biology. 41 (6): 806–811. doi:10.1079/IVP2005711. JSTOR4293939. S2CID26058596.
^ Károly N (2018). Immunohistochemical investigations of the neuronal changes induced by chronic recurrent seizures in a pilocarpine rodent model of temporal lobe epilepsy (Thesis). University of Szeged. doi:10.14232/phd.9734.
Last summer, Andrew Badrot bought a portfolio of plant-sourced pharmaceutical chemicals from Boehringer Ingelheim and acquired BI’s distribution rights for pilocarpine, a plant-derived glaucoma treatment.
For BI, the transactions were small ones. The German drugmaker had been exiting its private-label active pharmaceutical ingredients (API) business, scaling back to produce only the chemicals it uses to manufacture its own drugs.
But for Badrot the deals were potentially big. He leads the company that bought the businesses—Centroflora CMS, a joint venture between the Brazilian botanicals firm Centroflora and CMS Pharma, Badrot’s custom chemicals consultancy. Together, Centroflora and Centroflora CMS are committed to nurturing the natural source of pilocarpine, an alkaloid used medicinally for more than 100 years, and to expanding into other APIs neglected by larger firms.
Pilocarpine’s source, Pilocarpus microphyllus, better known as jaborandi, had been harvested vigorously in the wild by Merck KGaA, which in 1975 built a factory in Parnaíba in northern Brazil to extract pilocarpine. By the mid-1980s, however, jaborandi had been overharvested, and the government declared it a protected species. Merck began obtaining the leaves from a plantation in the northern Brazilian state of Maranhão.
Demand for the drug as a glaucoma treatment began to decline, and Merck eventually closed the plant. When the market for the drug revived with new indications as a dry-mouth remedy, the company saw an opportunity to sell the site and did so in 2002.
The buyer was Centroflora, which was founded in 1957 in São Paulo. The firm was interested in adding pilocarpine to its botanical extracts business, according to its chief executive, Peter Andersen, a native of Brazil whose coffee-trader father bought into Centroflora in 1983. Along with the purchase, Centroflora signed a deal for BI to distribute the drug.
The company wanted to revitalize natural harvesting of jaborandi and began working with the Brazilian government to promulgate sustainable practices in the field. Centroflora also worked closely with a German government agency, Deutsche Gesellschaft für Internationale Zusammenarbeit (GIZ), which promotes sustainable harvesting internationally and had been working in the north of Brazil for decades.
Centroflora’s distribution agreement with BI arose through connections at GIZ, according to Andersen. BI also had been Merck’s biggest customer for pilocarpine.
But ecological sustainability was only half of the problem, Andersen says. Centroflora also found itself dealing with middlemen who would collect the jaborandi from poor family farms in remote areas and pay them next to nothing. Establishing a direct supply channel was not easy.
“I can spend a few days telling you about that process,” he says. “Stories of difficult relationships and difficult moments. But in some cases we managed to hire some of the middlemen to work for us on a salary basis. They made less money, but they had a job.”
Today, farmers in Brazil are paid at least twice what they were paid by intermediaries, Andersen says.
Key to the process was a program Centroflora launched in 2004 called Partnerships for a Better World to train and certify growers, establish community associations to support growers, and maintain sustainable harvesting practices.
Centroflora is the leading supplier of pilocarpine. Its only competitor, Sourcetech, with a plant near São Paulo, accesses jaborandi from the plantation that supplied Merck, now owned by U.S.-based Quercegen.
Pilocarpine accounts for only about 5% of Centroflora’s $95 million in annual sales. The company produces a long list of botanical extracts, including nutritional supplements and herbal medicines such as acai, acerola, coffee powder, and powdered fruit.The company manufactures at four facilities in Brazil, including the former Merck plant, which is dedicated to pilocarpine. But Andersen sees the partnership with CMS as a route to increase phytochemical API manufacturing at that site.
“The facility has the capacity to produce 12 metric tons per year of alkaloids,” Andersen says. It currently makes less than three metric tons. “So there is a lot of space to produce more, and the idea is that we can do some of the APIs we got from Boehringer Ingelheim.”
Those include atropine, digoxin, homatropine, and dihydroergotamine mesylate. Centroflora CMS also obtained distribution rights to BI’s scopolamine N-butyl bromide. All are derived from botanicals harvested on farms around the world.
Badrot was vice president of strategy for Lonza’s exclusive synthesis division before starting CMS in 2010 to consult on manufacturing and mergers and acquisitions in the custom chemicals business. “But for me, the dream was to return to manufacturing APIs,” he says.
The phytochemicals portfolio, including some of the oldest APIs made by BI, for which CMS has done consulting work, seemed like an ideal reentry to manufacturing, according to Badrot. “They are niche products that maybe fly a bit under the radar,” he says. “They seemed to fit us well because we can give them some attention.”
Centroflora CMS’s first order of business, he says, is to establish manufacturing for the BI products, which BI will continue to make until then. Badrot says Centroflora is well suited to manufacture at least the digoxin and atropine, but decisions have not been finalized. The partners will likely use contract manufacturers for some of the products. And Badrot says Centroflora CMS seeks to replicate the kind of deal it has with BI.
“We are looking for other companies with APIs that represent 0–1% of sales, products that lack focus,” he says. “We would take them over.”
Badrot and Andersen say they are also interested in sharing the Partnerships for a Better World program with other companies involved in harvesting natural products. And Centroflora looks for other ways to support its supply chain. Last month, it was approved as a trading member of the Union for Ethical BioTrade, a nonprofit that promotes sustainable development and biodiversity. As a member, Centroflora commits to sustainable sourcing practices and will be required to undergo periodic audits.
Last year, Centroflora received government recognition for its efforts on both the environmental and social fronts. The National Confederation of Industry in Brazil named Centroflora’s jaborandi harvesting program one of the country’s 10 most sustainable business practices. And Banco do Brasil, the national bank, recognized the firm for its work to improve conditions for farmers in the northern forest region of the country.
As the joint venture starts to work with its new portfolio of phytochemicals, both Andersen and Badrot look back at the jaborandi success as the road forward, a template for fostering a plant-based API business that may inspire other companies.
For Andersen, Partnerships for a Better World is an essential foundation of trust for the ecological and socially responsible harvesting of botanicals in Brazil. “There were a lot of problems along the way,” he says. “But we are at peace with it today.”
CAS number 25655-41-8, Molecular Formula, (C6-H9-N-O)x-.x-I2, Molecular Weight, 364.9431
1-ethenylpyrrolidin-2-one;molecular iodine Povidone-Iodine CAS Registry Number: 25655-41-8 CAS Name: 1-Ethenyl-2-pyrrolidinone homopolymer compd with iodine Additional Names: 1-vinyl-2-pyrrolidinone polymers, iodine complex; iodine-polyvinylpyrrolidone complex; polyvinylpyrrolidone-iodine complex; PVP-I Trademarks: Betadine (Purdue Frederick); Betaisodona (Mundipharma); Braunol (Braun Melsungen); Braunosan H (Braun Melsungen); Disadine D.P. (Stuart); Efodine (Fougera); Inadine (J & J); Isodine (Blair); Proviodine (Rougier); Traumasept (Wolff) Literature References: An iodophor, q.v., prepd by Beller, Hosmer, US2706701; Hosmer, US2826532; Siggia, US2900305 (1955, 1958, and 1959, all to GAF). Prepn, history and use: Shelanski, Shelanski, J. Int. Coll. Surg.25, 727 (1956). Properties: Yellowish-brown, amorphous powder with slight characteristic odor. Aq solns have a pH near 2 and may be made more neutral (but less stable) by the addition of sodium bicarbonate. Sol in alc, water. Practically insol in chloroform, carbon tetrachloride, ether, solvent hexane, acetone. Solns do not give the familiar starch test when freshly prepared. Therap-Cat: Anti-infective (topical). Therap-Cat-Vet: Anti-infective (topical). Keywords: Antiseptic/Disinfectant; Halogens/Halogen Containing Compounds.
An iodinated polyvinyl polymer used as topical antiseptic in surgery and for skin and mucous membrane infections, also as aerosol. The iodine may be radiolabeled for research purposes.
Povidone-iodine is a stable chemical complex of polyvinylpyrrolidone (povidone, PVP) and elemental iodine. It contains from 9.0% to 12.0% available iodine, calculated on a dry basis. This unique complex was discovered in 1955 at the Industrial Toxicology Laboratories in Philadelphia by H. A. Shelanski and M. V. Shelanski. During in vitro testing to demonstrate anti-bacterial activity it was found that the complex was less toxic in mice than tincture of iodine. Human clinical trials showed the product to be superior to other iodine formulations. Povidone-iodine was immediately marketed, and has since become the universally preferred iodine antiseptic.
Povidone-iodine (PVP-I), also known as iodopovidone, is an antiseptic used for skin disinfection before and after surgery.[1][2] It may be used both to disinfect the hands of healthcare providers and the skin of the person they are caring for.[2] It may also be used for minor wounds.[2] It may be applied to the skin as a liquid or a powder.[2]
Wound area covered in povidone-iodine. Gauze has also been applied.
Povidone-iodine is a broad spectrum antiseptic for topical application in the treatment and prevention of wound infection. It may be used in first aid for minor cuts, burns, abrasions and blisters. Povidone-iodine exhibits longer lasting antiseptic effects than tincture of iodine, due to its slow absorption via soft tissue, making it the choice for longer surgeries. Chlorhexidine provides superior results with equivalent adverse events.[7]
Consequently, PVP-I has found broad application in medicine as a surgical scrub; for pre- and post-operative skin cleansing; for the treatment and prevention of infections in wounds, ulcers, cuts and burns; for the treatment of infections in decubitus ulcers and stasis ulcers; in gynecology for vaginitis associated with candidal, trichomonal or mixed infections. For these purposes PVP-I has been formulated at concentrations of 7.5–10.0% in solution, spray, surgical scrub, ointment, and swab dosage forms; however, use of 10% povidone-iodine though recommended, is infrequently used, as it is poorly accepted by health care workers and is excessively slow to dry.[8][9]
Because of these critical indications, only sterile povidone-iodine should be used in most cases. Non-sterile product can be appropriate in limited circumstances in which people have intact, healthy skin that will not be compromised or cut. The non-sterile form of Povidone iodine has a long history of intrinsic contamination with Burkholderia cepacia (aka Pseudomonas cepacia), and other opportunistic pathogens. Its ability to harbor such microbes further underscores the importance of using sterile products in any clinical setting. Since these bacteria are resistant to povidone iodine, statements that bacteria do not develop resistance to PVP-I,[10] should be regarded with great caution: some bacteria are intrinsically resistant to a range of biocides including povidone-iodine.[11]
Antiseptic activity of PVP-I is because of free iodine (I2) and PVP-I only acts as carrier of I2 to the target cells. Most commonly used 10% PVP-I delivers about 1-3 ppm of I2 in a compound of more than 31,600 ppm of total iodine atoms. All the toxic and staining effects of PVP-I is due to the inactive iodine only.
Eyes
A buffered PVP-I solution of 2.5% concentration can be used for prevention of neonatal conjunctivitis, especially if it is caused by Neisseria gonorrhoeae, or Chlamydia trachomatis. It is currently unclear whether PVP-I is more effective in reducing the number of cases of conjunctivitis in neonates over other methods.[12] PVP-I appears to be very suitable for this purpose because, unlike other substances, it is also efficient against fungi and viruses (including HIV and Herpes simplex).[13]
Pleurodesis
It is used in pleurodesis (fusion of the pleura because of incessant pleural effusions). For this purpose, povidone-iodine is equally effective and safe as talc, and may be preferred because of easy availability and low cost.[14]
Alternatives
There is strong evidence that chlorhexidine and denatured alcohol used to clean skin prior to surgery is better than any formulation of povidone-iodine[7]
The iodine in PVP-I reacts with hydrogen peroxide, silver, taurolidine and proteins such as enzymes, rendering them (and itself) ineffective. It also reacts with many mercury compounds, giving the corrosive compound mercury iodide, as well as with many metals, making it unsuitable for disinfecting metal piercings.[15]
Iodine is absorbed into the body to various degrees, depending on application area and condition of the skin. As such, it interacts with diagnostic tests of the thyroid gland such as radioiodine diagnostics, as well as with various diagnostic agents used on the urine and stool, for example Guaiacum resin.[15]
Free iodine, slowly liberated from the povidone-iodine (PVP-I) complex in solution, kills cells through iodination of lipids and oxidation of cytoplasmic and membrane compounds. This agent exhibits a broad range of microbiocidal activity against bacteria, fungi, protozoa, and viruses. Slow release of iodine from the PVP-I complex in solution minimizes iodine toxicity towards mammalian cells.
PVP-I can be loaded into hydrogels, which can be based on carboxymethyl cellulose (CMC), poly(vinyl alcohol) (PVA), and gelatin, or on crosslinked polyacrylamide. These hydrogels can be used for wound dressing. The rate of release of the iodine in the PVP-I is heavily dependent on the hydrogel composition: it increases with more CMC/PVA and decreases with more gelatin.
History
PVP-I was discovered in 1955, at the Industrial Toxicology Laboratories in Philadelphia by H. A. Shelanski and M. V. Shelanski.[18] They carried out tests in vitro to demonstrate anti-bacterial activity, and found that the complex was less toxic in mice than tincture of iodine. Human clinical trials showed the product to be superior to other iodine formulations.[19]
Following the discovery of iodine by Bernard Courtois in 1811, it has been broadly used for the prevention and treatment of skin infections, as well as the treatment of wounds. Iodine has been recognized as an effective broad-spectrum bactericide, and is also effective against yeasts, molds, fungi, viruses, and protozoans. Drawbacks to its use in the form of aqueous solutions include irritation at the site of application, toxicity, and the staining of surrounding tissues. These deficiencies were overcome by the discovery and use of PVP-I, in which the iodine is carried in a complexed form and the concentration of free iodine is very low. The product thus serves as an iodophor.
Research
Schematic of povidone-iodine complex wrapping a single wall carbon nanotube (black).[20]
Povidone-iodine has found application in the field of nanomaterials. A wound-healing application has been developed which employs a mat of single wall carbon nanotubes (SWNTs) coated in a monolayer of povidone-iodine.[20]
Research has previously found that the polymer polyvinylpyrrolidone (PVP, povidone) can coil around individual carbon nanotubes to make them water-soluble.[21]
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06.
^ Slater K, Cooke M, Fullerton F, Whitby M, Hay J, Lingard S, et al. (September 2020). “Peripheral intravenous catheter needleless connector decontamination study-Randomized controlled trial”. American Journal of Infection Control. 48 (9): 1013–1018. doi:10.1016/j.ajic.2019.11.030. PMID31928890.
^ Slater K, Fullerton F, Cooke M, Snell S, Rickard CM (September 2018). “Needleless connector drying time-how long does it take?”. American Journal of Infection Control. 46 (9): 1080–1081. doi:10.1016/j.ajic.2018.05.007. PMID29880433. S2CID46968733.
^ Fleischer W, Reimer K (1997). “Povidone-iodine in antisepsis–state of the art”. Dermatology. 195 Suppl 2 (Suppl 2): 3–9. doi:10.1159/000246022. PMID9403248.
^ Najafi Bi R, Samani SM, Pishva N, Moheimani F (2003). “Formulation and Clinical Evaluation of Povidone-Iodine Ophthalmic Drop”. Iranian Journal of Pharmaceuticical Research. 2 (3): 157–160.
^ Jump up to:abc Jasek W, ed. (2007). Austria-Codex (in German) (62nd ed.). Vienna: Österreichischer Apothekerverlag. pp. 983–5. ISBN978-3-85200-181-4.
^ Niedner R (1997). “Cytotoxicity and sensitization of povidone-iodine and other frequently used anti-infective agents”. Dermatology. 195 Suppl 2 (Suppl 2): 89–92. doi:10.1159/000246038. PMID9403263.
Wong RH, Hung EC, Wong VW, Wan IY, Ng CS, Wan S, Underwood MJ (2009). “Povidone-iodine wound irrigation: A word of caution”. Surgical Practice. 13 (4): 123–4. doi:10.1111/j.1744-1633.2009.00461.x. S2CID71797553.
Wong RH, Wong VW, Hung EC, Lee PY, Ng CS, Wan IY, Underwood MJ (2011). “Topical application of povidone-iodine before wound closure is associated with significant increase in serum iodine level”. Surgical Practice. 19 (3): 79–82. doi:10.1111/j.1744-1633.2011.00547.x. S2CID70528331.
5′-O-[Hydroxy({[2-(trimethylammonio)ethoxy]phosphinato}oxy)phosphoryl]cytidine 987-78-0[RN] 1-{5-O-[({Hydroxy[2-(trimethylammonio)ethoxy]phosphoryl}oxy)phosphinato]-β-D-ribofuranosyl}-4-imino-1,4-dihydro-2-pyrimidinol 213-580-7[EINECS], 2290 2-Pyrimidinol, 1,4-dihydro-1-[5-O-[hydroxy[[hydroxy[2-(trimethylammonio)ethoxy]phosphinyl]oxy]phosphinyl]-β-D-ribofuranosyl]-4-imino-, inner salt CiticolineCAS Registry Number: 987-78-0 CAS Name: Cytidine 5¢-(trihydrogen diphosphate) P¢-[2-(trimethylammonio)ethyl] ester inner salt Additional Names: choline cytidine 5¢-pyrophosphate (ester); cytidine diphosphate choline ester; CDP-choline Trademarks: Difosfocin (Magis); Nicholin (Wyeth); Recognan (Asahi); Rexort (Hoechst); Somazina (Ferrer) Molecular Formula: C14H26N4O11P2, Molecular Weight: 488.32 Percent Composition: C 34.43%, H 5.37%, N 11.47%, O 36.04%, P 12.69% Literature References: Naturally occurring nucleotide; intermediate in the major pathway of lecithin biosynthesis. Identification: E. P. Kennedy, S. B. Weiss, J. Am. Chem. Soc.77, 250 (1955).Crystallization from yeast extract: I. Lieberman et al.,Science124, 81 (1956).Synthesis: E. P. Kennedy, J. Biol. Chem.222, 185 (1956); K. Kikugawa et al.,Chem. Pharm. Bull.19, 1011, 2466 (1971). Molecular structure: M. A. Viswamitra et al.,Nature258, 497 (1975). Series of articles on pharmacology and toxicology: Arzneim.-Forsch.33, 1009-1080 (1983). Acute toxicity: T. Grau et al.,ibid. 1033. Clinical trial in ischemic stroke: W. M. Clark et al.,Neurology49, 671 (1997).Review of biosynthesis, metabolism, pharmacology: G. B. Weiss, Life Sci.56, 637-660 (1995); and clinical experience: J. J. Secades, G. Frontera, Methods Find. Exp. Clin. Pharmacol.17, Suppl. B, 1-54 (1995).Properties: Amorphous, somewhat hygroscopic powder. [a]D25 +19.0° (c = 0.86 in H2O). uv max (pH 1): 280 nm (e 12800). Dissolves readily in water to form acidic soln. Practically insol in most organic solvents. pKa 4.4. LD50 in mice, rats (mg/kg): 4600 ±335, 4150 ±370 i.v.; both species 8 g/kg orally (Grau). pKa: pKa 4.4Optical Rotation: [a]D25 +19.0° (c = 0.86 in H2O) Absorption maximum: uv max (pH 1): 280 nm (e 12800) Toxicity data: LD50 in mice, rats (mg/kg): 4600 ±335, 4150 ±370 i.v.; both species 8 g/kg orally (Grau) Derivative Type: Sodium saltCAS Registry Number: 33818-15-4 Trademarks: Acticolin (Upsamedica); Brassel (Searle); Ceraxon (Ferrer); Neuroton (Berlin-Chemie); Sintoclar (Pulitzer) Molecular Formula: C14H25N4NaO11P2, Molecular Weight: 510.31 Percent Composition: C 32.95%, H 4.94%, N 10.98%, Na 4.51%, O 34.49%, P 12.14% Properties: White, crystalline, spongy, hygrosopic powder, dec 250°. [a]D20 +12.5° (c = 1.0 in H2O). Sol in water. Practically insol in alcohol. Optical Rotation: [a]D20 +12.5° (c = 1.0 in H2O) Therap-Cat: Neuroprotective. In treatment of ischemic stroke and head trauma. Keywords: Neuroprotective.
Citicoline (INN), also known as cytidine diphosphate-choline (CDP-Choline) or cytidine 5′-diphosphocholine is an intermediate in the generation of phosphatidylcholine from choline, a common biochemical process in cell membranes. Citicoline is naturally occurring in the cells of human and animal tissue, in particular the organs.
Studies suggest that CDP-choline supplements increase dopamine receptor densities.[1] Intracerebroventricular administration of citicoline has also been shown to elevate ACTH independently from CRH levels and to amplify the release of other HPA axis hormones such as LH, FSH, GH and TSH in response to hypothalamic releasing factors.[2] These effects on HPA hormone levels may be beneficial for some individuals but may have undesirable effects in those with medical conditions featuring ACTH or cortisol hypersecretion including PCOS, type II diabetes and major depressive disorder.[3][4]
Citicoline was originally developed in Japan for stroke. Citicoline or its sodium salt was later introduced as a prescription drug in many European countries. In these countries it is now frequently prescribed for thinking problems related to circulation problems in the brain. In the US, citicoline is marketed as a dietary supplement. Citicoline or its sodium salt is used for Alzheimers disease and other types of dementia, head trauma, cerebrovascular disease such as stroke, age-related memory loss, Parkinsons disease, and glaucoma.
Citicoline sodium is chemically known as 5-0-[hydroxy({hydroxy[2-(trimethylammonio)ethoxy]phosphoryl}oxy)phosphoryl]cytidine sodium which is represented by formula I,
There are many process described in the art for the preparation of citicoline. Japanese patent 51028636 describes a process for the preparation of citicoline by neutralisation of Calcium salt of phosphorylcholine chloride with 98% H2SO4 to make phosphorylcholine chloride, which is further treated with cytidine-5-phosphate in presence of DCC and pyridine at 70 C to obtain citicoline hydrate. The drawback of this process is that citicoline is very unstable
in this harsh reaction condition such as formamide, 98% H2SO4 and high temperature of 70 C.
Chinese patent 1944661 describes an enzymatic process for the preparation of citicoline which involves twice pH adjustment to precipitatethe product,filtration of the solids, charcolisation, washing with pure water, eluting through chloride type ion exchange resin with water ethanol/alcohol reagents, desalting the eluate, decoloring and collecting the liquid, vacuum-concentration of the eluate by adding an alcohol solvent to get the solid to obtain the crude product and dissolving the crude product, microfiltering, ultrafiltering, adding an alcoholic solvent, to obtain the wet productand drying to obtain the final product. The primary disadvantage of this process is that the above reaction involves water and ethanol mixture for elution of ion exchange column and also vacuum concentration of water ethanol mixture which requires high energy, more time, leads to decomposition of product and also leads to the formation of more effluent hence it is not suitable for large scale production.
The primary disadvantage of this process is that the above reaction involves water and ethanol mixture for elution of ion exchange column and also vacuum concentration of water ethanol mixture which requires high energy, more time, leads to decomposition of product and also leads to the formation of more effluent hence it is not suitable for large scale production.
US20090286284 describes a microbial process for preparation of citicoline. This patent also discloses a process for purification of citicoline by passing through acidic cation exchange and anion exchange resin. The drawback of this process is that in this process citicoline is passed through cation /anion exchange resin in free form which is unstable and liable to formation of unwanted impurities. Therefore for the purification it needs very high volume of resin (200 times) and high volume (100 times) of solvent. This process further needs reconcentration of huge volume of solvents, which is time taking and energy consuming.
Chemical and Pharmaceutical Bulletin 1971, 19(5), 1011-16 describes a process for the preparation of citicoline by direct condensation of cytidine 5-
monophosphate and choline phosphate by using p-toluenesulfonyl chloride or methanesulfonyl chloride combined with DMF. After completion of reaction the mass was diluted with water, pH was adjusted with ammonia solution to 9.5 and product was purified by using Dowex-1 ion exchange resin by eluting with formic acid. Another Chemical and Pharmaceutical Bulletin 1971, 19(12), 2499-71 describes a process for the preparation of citicoline by direct condensation cytidine 5-monophosphate and choline phosphate in presence of thionyl chloride and DMF.The product obtained was further purified by using Dowex-1 ion exchange resin by eluting with formic acid.
Journal of Biological Chemistry, 1956, 185-191 describes a process for the preparation of citicoline by direct condensation5-cytidylic acid and phosphorylcholine in a mixture of water and pyridine in presence of DCC, stirred for few days by adding DCC in lots, after completion of reaction, reaction mass was diluted with water and filtered. The pH of the filtrate was adjusted 8-9 using 0.5N KOH, diluted further with water and passed through Dowex-1 formate resin by eluting with formic acid and water.
The drawbacks of these processes are that they use hazardous reagents such as p-toluenesulfonyl chloride, methanesulfonyl chloride, thionyl chloride etc. Hence they are not suitable for large scale production. Also, the prior art processes pass citicoline solution, without isolating it, to ion exchange resins for purification. During this process most of the inorganic impurities present along with citicoline or its salt pass through the column, thus making purification difficult.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
SYN
Journal of Chemical Research, 40(6), 358-360; 2016
An improved, three-step synthesis of cytidine diphosphate choline (CDP-choline) from cytidine was achieved in 68% overall yield. Selective phosphorylation of cytidine was accomplished by the use of morpholinophosphodichloridate to give cytidine-5′-phosphomorpholide, which was condensed with choline phosphate chloride in the presence of a catalytic amount of H2SO4 to give CDP-choline. The intermediates and products could be efficiently purified by recrystallisation, thus avoiding the use of chromatography at all stages. The reaction could be scaled up to 200 g in 64% overall yield, making this route attractive for industrial application.
Cytidine diphosphate choline (CDP-choline 1) is a nucleotide coenzyme and serves as a choline donor in the biosynthesis of lipids,1 lecithins,2 and sphingomyelin.3 It is a clinical drug for the treatment of several illnesses involving disturbance of the central nervous system, in particular, for regaining a patient’s consciousness and for treatment of neuropsychic symptoms occurring during skull traumas and brain surgery.4 Among various methods for the synthesis of CDP-choline in the literature, the current preferred method is via the condensation of cytidine-5’-phosphomorpholide (2) with choline phosphate chloride (3) under mild reaction conditions.5–7 Compound 2 was synthesised from 5’-cytidine monophosphate (4) and morpholine in the presence of DCC (N,N’-dicyclohexylcarbodiimide)8 or via the controlled hydrolysis of cytidine-5’-phosphodichloride (5) followed by P–N bond formation with morpholine (Scheme 1, route a).7 However, DCC is toxic and converted into urea which is difficult to separate from the mixture, thus leading to poor purity of product. Furthermore, phosphorylation with POCl3 always meets with side reactions from the 2’ or 3’ hydroxyls and detracts from the acceptance of this method in industry.9 In the context of ongoing projects on the synthesis of nucleoside drugs,10–14 herein we report the synthesis of CDP-choline via the selective phosphorylation of 6 wiResults and discussion Central to our approach for the synthesis of CDP-choline is the selective phosphorylation of 6 using sterically-hindered 7 as phosphorylation regent. 7 was synthesised by the direct phosphorylation of morpholine with POCl3 , a compound whose utility for the conversion of alcohols and amines into various phosphorylation derivatives.15 Due to the reactivity of three chloro atoms in POCl3 , gradually adding POCl3 to excess morpholine avoids the bifunctional reaction exclusively. After reaction, 7 could be purified by fractional distillation to yield as a colourless oil (b.p. 124–126 °C at 1.33 KPa). Due to the presence of the electron-donating morpholino group, 7 displays lower reactivity than POCl3 and could tolerate moisture and air better. Usefully, 7 could be synthesised on the 200 g scale and stored at 4 °C. The major concern of utilising 7 as phosphorylation reagent is its selectivity for the 5’ hydroxyl group. We therefore assessed the selectivity for 5’ hydroxylation using 6 and 7 in the presence of various organic bases. After phosphorylation, H2 O was added to destroy the excess of 7, and 2 was obtained in a single step. The solvent, the base, temperature and the ratio of substrates were evaluated and the results are summarised in Table 1
Cytidine-5’-phosphomorpholide (2): Cytidine (0.243 g, 1.0 mmol) and DMAP (0.183 g, 1.5 mmol) in MeCN (10 mL) were stirred slowly and cooled to 0 °C, and 7 (2.0 mmol) was added slowly. The mixture was heated to 50 °C and kept at this temperature for 2 h. The solvent was removed in vacuo and the residue was purified by recrystallisation from EtOH to give 2 as a white semi-solid (0.318 g); yield 81%; m.p. 62–64 °C; 1 H NMR (400 MHz, DMSO-d6 ) δ 8.43 (d, J = 7.6 Hz, 1H), 7.39 (s, 2H), 7.19 (d, J = 7.6 Hz, 1H), 5.77 (d, J = 2.8 Hz, 1H), 5.51 (d, J = 4.8 Hz, 1H), 5.18 (t, J = 5.2 Hz, 1H), 5.08 (d, J = 5.6 Hz, 1H), 3.76–3.71 (m, 1H), 3.61–3.56 (m, 1H), 3.45–3.42 (m, 4H), 3.03–2.99 (m, 4H); 13C NMR (100 MHz, DMSO-d6 ) δ 166.5, 157.8, 145.9, 141.6, 88.1, 86.2, 74.3, 70.7, 65.1, 65.0, 60.3, 60.2; HRMS calcd for C13H22N4 O8 P [M + H]+ 393.1170, found: 393.1172.
CDP-choline (1): 2 (0.392 g, 1.0 mmol) was added to MeOH (10 mL) followed by the addition of 3 (0.310 g, 1.2 mmol) and was stirred at room temperature for 10 min. Then 98% H2 SO4 (0.005 mL, 10 mol%) was added. The mixture was kept at 50 °C for 3 h. The solvent was removed in vacuo and the residue was purified by recrystallisation from EtOH to give 1 as a white solid (0.410 g); yield 85%. 1 H NMR (400 MHz, D2 O) δ 7.86 (s, 2H), 6.04 (d, J = 5.2 Hz, 1H), 5.91 (d, J = 5.2 Hz, 1H), 4.32 (brs, 2H), 4.26–4.22 (m, 2H), 4.18 (brs, 2H), 4.11 (t, J = 3.2 Hz, 1H), 3.60 (t, J = 2.4 Hz, 2H), 3.14 (s, 9H); 13C NMR (100 MHz, D2 O) δ 166.1, 157.7, 141.5, 96.6, 89.3, 82.6, 74.1, 69.3, 66.0, 65.9, 64.8, 59.9, 54.0; HRMS calcd for C14H27N4 O11P2 [M + H]+ 489.1146, found: 489.1140.
1H NMR
(400 MHz. D2O) δ 7.86 (s, 2H). 6.04 (d. J = 5.2 Hz, 111). 5.91 (d. J = 5.2 Hz. 1Hj, 4.32 (brs. 2H), 4.26-4.22 (m, 2H). 4.18 (brs, 2H), 4.11 (t. J = 3.2 Hz. 1H). 3.60 (t. J = 2.4 Hz. 2H), 3.14 (s, 9H)
calcd for C14H27N4O11P2 (M + H]+ 489.1146. found: 489.1140
State
white solid
SYNKikugawa, Kiyomi; Ichino, MotonobuChemical & Pharmaceutical Bulletin (1971), 19, (5), 1011-16.https://www.jstage.jst.go.jp/article/cpb1958/19/5/19_5_1011/_pdf/-char/enCytidine diphosphate choline (CDP-choline), one of the nucleotide coenzymes, is known to be a precursor of phospholipid and play an important role in the living organisms. The coenzyme was synthesized in a fairly good yield by direct condensation of cytidine-5′ monophosphate (5′-CMP) and choline phosphate (P-choline) by the use of p-toluenesulfonyl chloride or methanesulfonyl chloride combined with dimethylformamide Method B, with Methanesulfonyl Chloride and DMF: A mixture of 1.3g (11.5 mmole) of methanesulfonyl chloride and 3ml of DMF was added to the gummy mixture containing 10 mmole of P-choline (II). It was shaken at room temperature for 10 min, and 1.0g (3.1 mmole) of 5′-CMP (I) was added to the viscous solution. It was then stirred at room temperature for one hour. Paper chromatography and paper electrophoresis of the reaction mixture showed that CDP-choline (III) was a major reaction product. The separa tion, isolationand identification of the product (III) were same as in method A. Crystalline white powder of CDP-choline was obtained in a yield of 50.0%. Method C, with p-Toluenesulfonyl Chloride and HMPA: A mixture of 2.2g (11.5 mmole) of p-toluene sulfonyl chloride and 3ml of HMPA was added to the gummy mixture containing 10 mmole of P-choline (II). 5′-C1IP (I) (1.0g, 3.1 mmole) was reacted under the same condition as in method A, and isolation was performed similarly. Crystalline powder of CDP-choline (III) was obtained in a yield of about 10%. Method D according to the Morpholidate Method 6): 5′-CMP-Morpholidate (4-morpholine-N, N’-dicyclohexylcarboxamidinium salt) (1.28g, 2 mmole) was reacted with 8 mmole of P-choline (II) according to the method of Tanaka, et al. 6) Separation and isolation of the product were similarly performed as in method A. Crystalline powder of the authentic CDP-choline was obtained in a yield of 55%. CDP-Choline Monosodium Salt Monosodium salt of CDP-choline (III) was prepared from the product (III) obtained by method A. Thus, 200mg of CDP-choline (III) was dissolved in 1.0ml of water, and after the pH of the solution was adjusted to 6.0 with 2N NaOH, 3ml of ethanol was added. Crystallization occurred after standing at room temperature overnight to afford plates of 130 mg of CDP-choline monosodium salt. Determination of the Yield of CDP-Choline (III) in the Condensation with p-Toluenesulfonyl Chloride and DMF In the condensation reaction using p-toluenesulfonyl chloride and DMF, the effects of the amount of p-toluenesulfonyl chloride and the reaction temperature were examined. 5′-CMP (I) (1.0g, 3.1 mmole) was added to the mixture of 10 mmole of P-choline (II), 3ml of DMF and p-toluenesulfonyl chloride which were previously mixed and treated at room temperature for 10 min. The reaction mixtures were stirred at 25•‹for one hour with the varying amounts of p-toluenesulfonyl chloride of 1.5g (7.9 mmole), 1.9g (10 mmole), 2.2g (11.5 mmole), 3g (15.8 mmole), 4g (21.0 mmole) and 5g (26.3 mmole). The yields of the compound (III) estimated were 30, 50, 60, 49, 44 and 37% respectively.
SYN
Indian Pat. Appl., 2014MU00923
SYN
CN 111647636
Syn
Biotechnology and Bioengineering, 117(5), 1426-1435; 2020
Cytidine-5′-diphosphocholine (CDP-choline) is a widely used neuroprotective drug for multiple indications. In industry, CDP-choline is synthesized by a two-step cell culture/permeabilized cell biotransformation method because substrates often do not enter cells in an efficient manner. This study develops a novel one-step living cell fermentation method for CDP-choline production. For this purpose, the feasibility of Pichia pastoris as a chassis was demonstrated by substrate feeding and CDP-choline production. Overexpression of choline phosphate cytidylyltransferase and choline kinase enhanced the choline transformation pathway and improved the biosynthesis of CDP-choline. Furthermore, co-overexpression of ScHnm1, which is a heterologous choline transporter, highly improved the utilization of choline substrates, despite its easy degradation in cells. This strategy increased CDP-choline titer by 55-folds comparing with the wild-type (WT). Overexpression of cytidine-5′-monophosphate (CMP) kinase and CDP kinase in the CMP transformation pathway showed no positive effects. An increase in the ATP production by citrate stimulation or metabolic pathway modification further improved CDP-choline biosynthesis by 120%. Finally, the orthogonal optimization of key substrates and pH was carried out, and the resulting CDP-choline titer (6.0 g/L) at optimum conditions increased 88 times the original titer in the WT. This study provides a new paradigm for CDP-choline bioproduction by living cells.
SYN
Citicoline sodium is a chemically designate as Cytidine 5’-(trihydrogendiphosphate) P’-[2-(trimethylammonio) ethyl] ester monosodium salt, its molecular formula is C14H25N4NaO11P2 and molecular weight is 510.31(salt) and 488.32 (base- C14H26N4O11P2). It is a white crystalline, hygroscopic powder and readily soluble in water but practically insoluble in alcohol. Its melting point was 259 – 268°C and dissociation constant (Pka) was 4.4 [1]. Biopharmaceutical classification system (BCS) for Citicoline is Class – I (High solubility and High Permeability) [3]. Citicoline has a broad spectrum of therapeutic index, as a Neuroprotectant or Cerebroprotectant, in particular citicoline is useful the victims of ischemic stroke, head trauma and neurodegenerative disease. Citicoline is also used to treat unconsciousness resulting from cerebral thrombosis, hemorrhages, demyelinating diseases, cranial trauma and cerebropathies due to atherosclerosis [2]. Citicoline was originally developed in Japan for stroke. It was later introduced as a prescription drug in many European countries. In these countries it is now frequently prescribed for thinking problems related to circulation problems in the brain. In the US, citicoline is marketed as a dietary supplement [3]. Citicoline daily dosages may range from 250 mg to about 3000 mg and more preferably from 500 mg to about 2000 mg up to four or more times daily, duration of the treatment may vary from several weeks to several years, dosages may be varied over time depending on the severity of symptoms [4].
SYN
192/MUM/2012
The present invention discloses a novel, cost-effective process for preparing psychostimulant drug cytidinediphosphate-choline (CDP-Choline) commonly known as citicoline. The process comprises reacting cytidine 5-monophosphate with morpholine in presence of a coupling reagent and an organic solvent to form morpholidate compound; condensing morpholidate compound with calcium salt of phosphorylcholinehalide in presence of an acid to form citicoline calcium chloride; and purifying the citicoline calcium chlorideby passing through cationic and anionic resinsand eluting by water to form citicoline sodium of formula I.
Example:
(a) Preparation of citicoline calcium chloride:
S-Cytidine mono phosphate (1.25 kg)and morpholine (1.12 kg) were added into methanol {6.25 L) and DCC (1.50 kg) at 25 to 35 C.The reaction mixture was heated to 50 to 55 C and stirred for 7 hours. After completion of the reaction, the reaction mass was cooled to 25 to 35 C and the obtained reaction mass was added slowly to phosphoryl choline chloride calcium salt (1.9 kg) in methanol (8.75 L) solution. The pH was maintained to 3.8 to 4.2 using HC1 gas in IPA and stirred for 6 hours at 25 to 35 . The reaction mass was further heated to 45 to 5Q C. After completion of reaction the yeactkm mass was cooled and stirced for 1 hour. The product was filtered, washed with chilled methanol at 0 to 5 Cand suck dried to obtain citicoline calcium chloride.
Yield; 3.70-4.0 kg (b) Preparation of citicoline sodium:
The above obtained crude citicolinecalcium chloride was dissolved in water (6.25 L), filtered, washed with water and suck dried. Filtrate containing the product was re-filtered through Hyflo bed. The clear filtrate was eluted through column containing acidic cation exchange resins (12.5 L). The material was washed with water. The eluent was further passed through anion exchange resin (12.5 L). column and washed with water.
Complete aqueous solution after the passing through an-ion exchange resin was collected, pH of the solution was adjusted to 6.5 to 7.0 using 30 % sodium hydroxide solution (0.3Kg in 0.45L) and solution was concentrated using reverse osmosis. The solution was cooled to 25 to 35 C and charcoalated. The solution was filtered through hyflo bed at 25 to 35 C, washed with water. The solution was further filtered through ultra-filter at 25 to 35 C.
Clear filtrate and mixture of isopropanol and Methanol (1:1) (25 L) were stirred, the reaction mass was cooled to 0 to 5 C, and stirred for 2 hours. The product was filtered under nitrogen atmosphere, solid was washed with the mixture of IPA and methanol (1:1) (1.25 L) at 0 to 5 C and dried under vacuum below 95 C until moisture/LOD is less the 2.0%.
https://patents.google.com/patent/CN1944661A/enEmbodiment 1:With 30 kilograms of quick-frozen yeast, 3 kilograms of phosphorylcholines, 1 kilogram 5 ‘-cytidylic acid, 10 kilograms of glucose, 2 kilograms of potassium hydroxide, 800 kg of water are mixed back temperature adjustment to 25 ℃, PH=6 carries out 65 rev/mins of stirring reactions and it was fully reacted in 6 hours; Reaction solution is warming up to 50 ℃ of deactivations, carries out liquid-solid separation; Transfer PH=8.0, part basic protein and nucleic acid precipitation are carried out liquid-solid separation, and then are transferred PH=2.5, make the acidic protein precipitation, carry out liquid-solid separation, sediment separate out; Use Activated Carbon Adsorption Separation, PH=2.5 washs with pure water; Carry out wash-out with the molten reagent of ethanol alkali, elutriant carries out desalination, decolouring is handled, and collects liquid; The elutriant vacuum concentration; Concentrated solution adds 2 times of ethanol, crystallization, liquid-solid separate crude product; Dissolving crude product, ultrafiltration behind the micro-filtration adds 2 times of ethanol, crystallization, liquid-solid separate wet product, after the drying finished product.Embodiment 2:With 80 kilograms of quick-frozen yeast, 4 kilograms of phosphorylcholines, 4 kilogram 5 ‘-cytidylic acid, 16 kilograms of glucose, 4 kilograms of potassium hydroxide, 1100 kg of water are mixed back temperature adjustment to 30 ℃, and add 0.5 kilogram of MgSO 4Solution, PH=6 carries out 120 rev/mins of stirring reactions and it was fully reacted in 8 hours; Reaction solution is warming up to 70 ℃ of deactivations, carries out liquid-solid separation; Transfer PH=10, part basic protein and nucleic acid precipitation are carried out liquid-solid separation, and then are transferred PH=4, make the acidic protein precipitation, carry out liquid-solid separation, sediment separate out; Use Activated Carbon Adsorption Separation, PH=4 washs with pure water; Carry out wash-out with the molten reagent of ethanol alkali, elutriant carries out desalination, decolouring is handled, and collects liquid; The elutriant vacuum concentration; Concentrated solution adds 2 times of methyl alcohol, crystallization, liquid-solid separate crude product; Dissolving crude product, ultrafiltration behind the micro-filtration adds 2 times of methyl alcohol, crystallization, liquid-solid separate wet product, after the drying finished product.Embodiment 3:With 100 kilograms of quick-frozen yeast, 8 kilograms of phosphorylcholines, 5 kilogram 5 ‘-cytidylic acid, 20 kilograms of glucose, 5 kilograms of potassium hydroxide, 1500 kg of water are mixed back temperature adjustment to 40 ℃, and add 6 kilograms of MgSO 4Solution, PH=8 carries out 150 rev/mins of stirring reactions and it was fully reacted in 10 hours; Reaction solution is warming up to 90 ℃ of deactivations, carries out liquid-solid separation; Transfer PH=12.0, part basic protein and nucleic acid precipitation are carried out liquid-solid separation, and then are transferred PH=5.5, make the acidic protein precipitation, carry out liquid-solid separation, sediment separate out; Use Activated Carbon Adsorption Separation, PH=5.5 washs with pure water; Carry out wash-out with the molten reagent of ethanol alkali, elutriant carries out desalination, decolouring is handled, and collects liquid; The elutriant vacuum concentration; Concentrated solution adds 2 times of first and second alcoholic solution, crystallization, liquid-solid separate crude product; Dissolving crude product, ultrafiltration behind the micro-filtration adds 2 times of first and second alcoholic solution, crystallization, liquid-solid separate wet product, after the drying finished product. PATENThttps://patents.google.com/patent/WO2013128393A1/enCiticoline (CDP-Choline), naturally occurring nucleotide, is a neuroprotective indicated for the treatment of ischemic stroke and head trauma in patients. Citicoline (CDP-Choline) is represented by formula (I).
US patent no. 3,666,748 discloses a process for preparing Citicoline sodium by reaction of 4- morpholino-N,N’-dicyclohexylcarboxamidine chloride salt of choline phosphormorpholidate (I) with cytidine-5-monophosphate in free form or its salts with base in a solvent such as o- chlorophenol, m-cresol, acetonitrile, pyridine and the like. The Citicoline thus obtained is purified through a column chromatograph packed with activated carbon followed by elution to get ammonium salt of citicoline, which is further converted to citicoline followed by citicoline sodium.US patent no. 3,787,392 discloses a process for preparing Citicoline by adding the acidic calcium phosporyl choline chloride tetra hydrate to the solution of morpholidiate cytidine 5- monophosphate and DCC in methanol followed by isolation and purification by means of chromatography column containing anion exchanger (Dowex 1×2 type formate form; 50-100 mesh) which is further converted to its sodium salt by neutralizing with sodium hydroxide. Further, US patent no. 3,803,125 discloses a process for preparing citicoline by reacting morpholidiate cytidine 5 ‘-monophosphate with calcium phosporyl choline chloride tetra hydrate in solvent system of an aliphatic alcohol or dialkyl ketone or dimethyl formamide at pH from 1 to 6.5. The product thus obtained is further isolated; purified by means of chromatography column containing anion exchanger; concentrated; and neutralized with aqueous solution of sodium hydroxide to get citicoline sodium.Example 1To a solution of calcium phosphoryl choline chloride tetra hydrate (50.0 gm) in water, a solution of oxalic acid in RO water (19.5 gm oxalic acid in 90 ml RO water) was added at 45- 50°C. The reaction mass was filtered and distilled out to get residue followed by addition of methanol. To the above solution, solution of morpholine and DCC in methanol was added. The temperature of the reaction was raised to 50-55°C and to this, solution of cytidine 5′- monophospahte in methanol (12.2 gm in 40 ml methanolic HCl and 20 ml methanol) was added and reaction was maintained. The pH 3.5 of reaction mixture was maintained by methanolic HCl. Reaction mass was cooled and IPA was added after completion of the reaction. The precipitated product, citicoline, was filtered and dried. The crude Citicoline (16.0 gm) was dissolved in water and treated with charcoal to get the purified Citicoline acid which on reaction with aqueous sodium hydroxide gave Citicoline Sodium with purity > 99%.Example 2To the solution of cytidine 5′-monophospahate (5′-CMP) (100 gm) in methanol (750 ml), solution of morpholine (75 gm) and DCC (100 gm) in methanol was added at room temperature. The temperature of the reaction was raised to 50-55°C for a time period of 3-7 hrs followed by cooling the reaction mass and filtered to get morpholidiate cytidine 5’- monophospahate in mother liquor. To this, solution of calcium phosphoryl choline chloride (200 gm) in methanol was added and the temperature of reaction mass was raised to 50-55°C and maintained at pH of 3.5 by methanolic HCl. The reaction mass was cooled and filtered to get crude Citicoline by adding IPA. Further, morpholidiate salt of oxalic acid (138.3 gm) was added to the solution of crude citicoline in methanol at 30-35°C followed by the addition of IPA to get the precipitated Citicoline, which is further treated with activated charcoal in water followed by filtration. To filtrate containing purified Citicoline, aqueous solution of sodium hydroxide was added at room temperature followed by addition of ethanol and the temperature of reaction mass was raised to 50-55°C. The precipitated product was filtered and dried where the purity of citicoline sodium is > 99% measured by HPLC. (265 gm). ClaimsHide Dependent
We Claim:1. A process for preparing pure Citicoline (CDP-Choline), the process comprising:reacting a cytidine 5′-monophospahte or its amide salts with calcium phosphoryl choline chloride tetra hydrate or its amide salts in presence of dicyclohexyl carbodiimide (DCC) and a solvent,wherein a dicarboxylic acid or its salt is employed in the process to obtain citicoline with a purity of more than 99% measured by HPLC.2. The process as claimed in claim 1, further comprising preparing highly pure sodium salt of citicoline by reacting the pure citicoline with sodium hydroxide.3. The process as claimed in claim 1, wherein the dicarboxylic acid is used either in the form of free acid or its base salts.4. The process as claimed in any one of the preceding claims, wherein dicarboxylic acid is selected from the group consisting of oxalic acid, malonic acid, succininc acid and glutaric acid.5. The process as claimed in any one of the preceding claims, wherein the base of dicarboxylic acid is selected from the group consisting of organic bases such as amidates, amines or inorganic base such as alkali or alkaline earth metal.6. The process as claimed in claim 1, wherein the solvent is selected from the group consisting of aliphatic alcohols from C atoms, ketones such as acetone, methyl isobutyl ketone and the like or mixture thereof.7. The process as claimed in claim 1, wherein the solvent is methanol.8. The process as claimed in any of the preceding claims, wherein the dicarboxylic acid or its salts lessen the solubility of inorganic impurities such as calcium chloride, calcium hydroxide, unreacted choline phosphate, 5-CMP.
Patent
US3666748A *1967-12-181972-05-30Takeda Chemical Industries LtdMethod for production of cytidine (or deoxycytidine)-5{40 -diphosphate choline and intermediates thereforUS3787392A *1970-12-021974-01-22Boehringer Mannheim GmbhProcess for the preparation of nucleoside diphosphate estersFamily To Family CitationsCN102010454B *2010-12-022012-03-07胡建荣Citicoline sodium compound and new method thereofPublication numberPriority datePublication dateAssigneeTitleCN104031105A *2014-06-062014-09-10浙江天冉药物研究有限公司Method for preparing citicoline sodiumCN105732752A *2016-03-182016-07-06新乡学院Citicoline and synthetic method thereofCN106146590A *2016-06-292016-11-23陈建峰A kind of preparation method of C14H25N4NaO11P2CN110684066A *2019-05-222020-01-14广东金城金素制药有限公司Cytophosphocholine medicinal preparation and new application thereof in cerebral infarction acute-stage disturbance of consciousness
Citicoline is available as a supplement in over 70 countries under a variety of brand names: Cebroton, Ceraxon, Cidilin, Citifar, Cognizin, Difosfocin, Hipercol, NeurAxon, Nicholin, Sinkron, Somazina, Synapsine, Startonyl, Trausan, Xerenoos, etc.[5] When taken as a supplement, citicoline is hydrolyzed into choline and cytidine in the intestine.[6] Once these cross the blood–brain barrier it is reformed into citicoline by the rate-limiting enzyme in phosphatidylcholine synthesis, CTP-phosphocholine cytidylyltransferase.[7][8]
Research
Memory and cognition
Studies have failed to confirm any potential benefits of citicoline for cognitive impairment.[9]
Ischemic stroke
Some preliminary research suggested that citicoline may reduce the rates of death and disability following an ischemic stroke.[10][11] However, the largest citicoline clinical trial to date (a randomised, placebo-controlled, sequential trial of 2298 patients with moderate-to-severe acute ischaemic stroke in Europe), found no benefit of administering citicoline on survival or recovery from stroke.[12] A meta-analysis of seven trials reported no statistically significant benefit for long-term survival or recovery.[13]
Vision
The effect of citicoline on visual function has been studied in patients with glaucoma, with possible positive effect for protecting vision.[14]
Mechanism of action
Enzymes involved in reactions are identified by numbers. See file description.
By converting 1, 2-diacylglycerol into phosphatidylcholine
Stimulating the synthesis of SAMe, which aids in membrane stabilization and reduces levels of arachidonic acid. This is especially important after an ischemia, when arachidonic acid levels are elevated.[19]
Neuronal membrane
The brain preferentially uses choline to synthesize acetylcholine. This limits the amount of choline available to synthesize phosphatidylcholine. When the availability of choline is low or the need for acetylcholine increases, phospholipids containing choline can be catabolized from neuronal membranes. These phospholipids include sphingomyelin and phosphatidylcholine.[15] Supplementation with citicoline can increase the amount of choline available for acetylcholine synthesis and aid in rebuilding membrane phospholipid stores after depletion.[20] Citicoline decreases phospholipase stimulation. This can lower levels of hydroxyl radicals produced after an ischemia and prevent cardiolipin from being catabolized by phospholipase A2.[21][22] It can also work to restore cardiolipin levels in the inner mitochondrial membrane.[21]
Cell signalling
Citicoline enhances cellular communication by increasing the availability of neurotransmitters, including acetylcholine, norepinephrine, and dopamine.[23] In simple terms, the choline component of citicoline is used to create acetylcholine, which is a primary executive neurotransmitter in the human brain. Clinical trials have found that citicoline supplementation improves attention, focus and learning in large part due to the increase in acetylcholine that results.[24]
Glutamate transport
Citicoline lowers increased glutamate concentrations and raises decreased ATP concentrations induced by ischemia. Citicoline also increases glutamate uptake by increasing expression of EAAT2, a glutamate transporter, in vitro in rat astrocytes. It is suggested that the neuroprotective effects of citicoline after a stroke are due in part to citicoline’s ability to decrease levels of glutamate in the brain.[25]
Pharmacokinetics
Citicoline is water-soluble, with more than 90% oral bioavailability.[20] Plasma levels peak one hour after oral ingestion, and a majority of the citicoline is excreted as CO2 in respiration, and again 24 hours after ingestion, where the remaining citicoline is excreted through urine.[26]
Side effects
Citicoline has a very low toxicity profile in animals and humans. Clinically, doses of 2000 mg per day have been observed and approved. Minor transient adverse effects are rare and most commonly include stomach pain and diarrhea.[17][unreliable medical source?] There have been suggestions that chronic citicoline use may have adverse psychiatric effects. However, a meta-analysis of the relevant literature does not support this hypothesis.[27][28] At most, citicoline may exacerbate psychotic episodes or interact with anti-psychotic medication.
Synthesis
In vivo
Phosphatidylcholine is a major phospholipid in eukaryotic cell membranes. Close regulation of its biosynthesis, degradation, and distribution is essential to proper cell function. Phosphatidylcholine is synthesized in vivo by two pathways
^ Cavun S, Savci V (Oct 2004). “CDP-choline increases plasma ACTH and potentiates the stimulated release of GH, TSH and LH: the cholinergic involvement”. Fundamental & Clinical Pharmacology. 18 (5): 513–23. doi:10.1111/j.1472-8206.2004.00272.x. PMID15482372. S2CID33866107.
^ Benson S, Arck PC, Tan S, Hahn S, Mann K, Rifaie N, Janssen OE, Schedlowski M, Elsenbruch S (Jun 2009). “Disturbed stress responses in women with polycystic ovary syndrome”. Psychoneuroendocrinology. 34 (5): 727–35. doi:10.1016/j.psyneuen.2008.12.001. PMID19150179. S2CID13202703.
^ Florio P, Zatelli MC, Reis FM, degli Uberti EC, Petraglia F (2007). “Corticotropin releasing hormone: a diagnostic marker for behavioral and reproductive disorders?”. Frontiers in Bioscience. 12: 551–60. doi:10.2741/2081. PMID17127316.
^ Single-ingredient Preparations (: Citicoline). In: Martindale: The Complete Drug Reference [ed.by Sweetman S], 35th Ed. 2007, The Pharmaceutical Press: London (UK); e-version. .
^ Wurtman RJ, Regan M, Ulus I, Yu L (Oct 2000). “Effect of oral CDP-choline on plasma choline and uridine levels in humans”. Biochemical Pharmacology. 60 (7): 989–92. doi:10.1016/S0006-2952(00)00436-6. PMID10974208.
^ Alvarez XA, Sampedro C, Lozano R, Cacabelos R (Oct 1999). “Citicoline protects hippocampal neurons against apoptosis induced by brain beta-amyloid deposits plus cerebral hypoperfusion in rats”. Methods and Findings in Experimental and Clinical Pharmacology. 21 (8): 535–40. doi:10.1358/mf.1999.21.8.794835. PMID10599052.
^ Carlezon WA, Pliakas AM, Parow AM, Detke MJ, Cohen BM, Renshaw PF (Jun 2002). “Antidepressant-like effects of cytidine in the forced swim test in rats”. Biological Psychiatry. 51 (11): 882–9. doi:10.1016/s0006-3223(01)01344-0. PMID12022961. S2CID21170398.
^ Warach S, Pettigrew LC, Dashe JF, Pullicino P, Lefkowitz DM, Sabounjian L, Harnett K, Schwiderski U, Gammans R (Nov 2000). “Effect of citicoline on ischemic lesions as measured by diffusion-weighted magnetic resonance imaging. Citicoline 010 Investigators”. Annals of Neurology. 48 (5): 713–22. doi:10.1002/1531-8249(200011)48:5<713::aid-ana4>3.0.co;2-#. PMID11079534.
^ Saver JL (Fall 2008). “Citicoline: update on a promising and widely available agent for neuroprotection and neurorepair”. Reviews in Neurological Diseases. 5 (4): 167–77. PMID19122569.
^ Dávalos A, Alvarez-Sabín J, Castillo J, Díez-Tejedor E, Ferro J, Martínez-Vila E, Serena J, Segura T, Cruz VT, Masjuan J, Cobo E, Secades JJ (Jul 2012). “Citicoline in the treatment of acute ischaemic stroke: an international, randomised, multicentre, placebo-controlled study (ICTUS trial)”. Lancet. 380 (9839): 349–57. doi:10.1016/S0140-6736(12)60813-7. hdl:10400.10/663. PMID22691567. S2CID134947.
^ Shi PY, Zhou XC, Yin XX, Xu LL, Zhang XM, Bai HY (2016). “Early application of citicoline in the treatment of acute stroke: A meta-analysis of randomized controlled trials”. J. Huazhong Univ. Sci. Technol. Med. Sci. 36 (2): 270–7. doi:10.1007/s11596-016-1579-6. PMID27072975. S2CID25352343.
^ López-Coviella I, Agut J, Savci V, Ortiz JA, Wurtman RJ (Aug 1995). “Evidence that 5′-cytidinediphosphocholine can affect brain phospholipid composition by increasing choline and cytidine plasma levels”. Journal of Neurochemistry. 65 (2): 889–94. doi:10.1046/j.1471-4159.1995.65020889.x. PMID7616250. S2CID10184322.
^ Jump up to:ab Conant R, Schauss AG (Mar 2004). “Therapeutic applications of citicoline for stroke and cognitive dysfunction in the elderly: a review of the literature”. Alternative Medicine Review. 9 (1): 17–31. PMID15005642.
^ Babb SM, Wald LL, Cohen BM, Villafuerte RA, Gruber SA, Yurgelun-Todd DA, Renshaw PF (May 2002). “Chronic citicoline increases phosphodiesters in the brains of healthy older subjects: an in vivo phosphorus magnetic resonance spectroscopy study”. Psychopharmacology. 161 (3): 248–54. doi:10.1007/s00213-002-1045-y. PMID12021827. S2CID28454793.
Common side effects when used by mouth include itchiness and rash.[4] Common side effects when used by injection include vomiting and kidney problems.[4] While not recommended historically, starting allopurinol during an attack of gout appears to be safe.[5][6] In those already on the medication, it should be continued even during an acute gout attack.[5][3] While use during pregnancy does not appear to result in harm, this use has not been well studied.[1] Allopurinol is in the xanthine oxidase inhibitor family of medications.[4]
ALLUPRINOLCAS Registry Number: 315-30-0 CAS Name: 1,5-Dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one Additional Names: 1H-pyrazolo[3,4-d]pyrimidin-4-ol; 4-hydroxypyrazolo[3,4-d]pyrimidine; HPP Manufacturers’ Codes: BW-56158 Trademarks: Adenock (Mitsubishi); Allurit (Aventis); Aloral (Lagap); Alositol (Tanabe); Allo-Puren (Isis); Allozym (Sawai); Allural (Rovi); Anoprolin (Azwell); Anzief (Nippon Chemiphar); Apulonga (Dorsch); Apurol (Siegfried); Apurin (GEA); Bleminol (Gepepharm); Caplenal (Teva); Cellidrin (Hennig); Cosuric (DDSA); Dabroson (Hoyer); Embarin (Merckle); Epidropal (Teofarma); Foligan (DESMA); Gichtex (Gerot); Hamarin (Roche); Hexanurat (Durascan); Ketanrift (Ohta); Lopurin (Abbott); Lysuron (Roche); Miniplanor (Galen); Monarch (SS Pharm.); Remid (TAD); Riball (Schering AG); Sigapurol (Siegfried); Suspendol (Merckle); Takanarumin (Takata); Uricemil (Molteni); Uripurinol (Azupharma); Urosin (Roche); Urtias (Novartis); Zyloprim (GSK); Zyloric (GSK) Molecular Formula: C5H4N4O, Molecular Weight: 136.11 Percent Composition: C 44.12%, H 2.96%, N 41.16%, O 11.75% Literature References: Xanthine oxidase inhibitor; decreases uric acid production. Prepn: Robins, J. Am. Chem. Soc.78, 784 (1956); Schmidt, Druey, Helv. Chim. Acta39, 986 (1956); Druey, Schmidt, US2868803 (1959 to Ciba); GB798646 (1958 to Wellcome Found.); Hitchings, Falco, US3474098 (1969 to Burroughs Wellcome). Physiological and biochemical studies: Hitchings, in Biochem. Aspects Antimetab. Drug Hydroxylation, D. Shugar, Ed. (Academic Press, London, 1969) pp 11-22, C.A.75, 3531h (1971). Clinical trial in treatment of renal calculi: M. J. V. Smith, J. Urol.117, 690 (1977); B. Ettinger et al.,N. Engl. J. Med.315, 1386 (1986). Use in hyperuricemia and gout: G. R. Boss, J. E. Seegmiller, ibid.300, 1459 (1977). Effect on renal function in treatment of gout: T. Gibson, Ann. Rheum. Dis.41, 59 (1982). Comprehensive description: S. A. Benezra, T. R. Bennett, Anal. Profiles Drug Subs.7, 1-17 (1978). Properties: Crystals, mp above 350°. uv max (0.1N NaOH): 257 nm (e 7200); (0.1N HCl): 250 nm (e 7600); (methanol): 252 nm (e 7600). Soly in mg/ml at 25°: water 0.48; n-octanol <0.01; chloroform 0.60; ethanol 0.30; DMSO 4.6. pKa 10.2. Melting point: mp above 350° pKa: pKa 10.2 Absorption maximum: uv max (0.1N NaOH): 257 nm (e 7200); (0.1N HCl): 250 nm (e 7600); (methanol): 252 nm (e 7600) Derivative Type: Sodium salt CAS Registry Number: 17795-21-0 Trademarks: Aloprim (Nabi) Molecular Formula: C5H3N4NaO, Molecular Weight: 158.09Percent Composition: C 37.99%, H 1.91%, N 35.44%, Na 14.54%, O 10.12% Properties: White amorphous mass. pKa 9.31. pKa: pKa 9.31 Therap-Cat: Treatment of hyperuricemia and chronic gout. Antiurolithic. Keywords: Antigout; Antiurolithic; Xanthine Oxidase Inhibitor.
Synthesis ReferenceDruey, J. and Schmidt, P.; US. Patent 2868,803; January 13,1959; assigned to Ciba Pharmaceutical Products Inc. Hitchings, G.H. and Falco, EA.; U.S. Patent 3,474,098; October 21,1969; assigned to Bur- roughs Wellcome & Co. Cresswell, R.M.and Mentha, J.W.; US.Patent4,146,713; March27,1979; assigned to Bur- roughs Wellcome & Co. SYN
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
US 2 868 803 (Ciba; 13.1.1959; CH-prior. 10.2.1956).
DAS 1 720 024 (Wellcome Found; appl. 12.7.1967; GB-prior. 14.7.1966).
Similar process:
DAS 1 904 894 (Wellcome Found; appl. 31.1.1969; GB-prior. 2.2.1968).
US 4 146 713 (Burroughs Wellcome; 27.3.1979; GB-prior. 2.2.1968).
Alternative syntheses:
US 3 474 098 (Burroughs Wellcome; 21.10.1969; prior. 29.3.1956).
DAS 2 224 382 (Henning Berlin; appl. 18.5.1972).
DE 1 118 221 (Wellcome Found; appl. 4.8.1956; GB-prior. 10.8.1955).
DAS 1 814 082 (Wellcome Found; appl. 11.12.1968).
DAS 1 950 075 (Henning Berlin; appl. 3.10.1969).
SYNCondensation of hydrazine with ethoxymethylenemalononitrile (I) leads to 3-amino-4-cyanopyrazole (II), which, by hydrolysis with sulphuric acid, gives the corresponding amide (III); heating III with formamide in excess results in allopurinol (IV). The synthesis of allopurinol can be illustrated as below:
Infrared Spectrum The infrared spectrum of allopurinol is shown in Figure 1 . in KBr with a Perkin Elmer model 457 infrared spectrophotometer. with the structure of allopurinol . It was taken as a 0.2% dispersion of allopurinol Table I gives the infrareg assignments consistent Table I Infrared Spectral Assignments for Allopurinol Frequency (cm-l) Assignment
3060 CH stretching vibrations of the pyrimidine ring
1700 CO stretching vibration of the keto form of the 4-hydroxy tautomer 1
Allopurinol was also commonly used to treat tumor lysis syndrome in chemotherapeutic treatments, as these regimens can rapidly produce severe acute hyperuricemia;[10] however, it has gradually been replaced by urate oxidase therapy.[11]Intravenous formulations are used in this indication when people cannot take medicine by mouth.[12]
Inflammatory bowel disease
Allopurinol cotherapy is used to improve outcomes for people with inflammatory bowel disease and Crohn’s disease who do not respond to thiopurine monotherapy.[13][14] Cotherapy has also been shown to greatly improve hepatoxicity side effects in treatment of IBD.[15] Cotherapy invariably requires dose reduction of the thiopurine, usually to one-third of the standard dose depending upon the patient’s genetic status for thiopurine methyltransferase.[16]
Psychiatric disorders
Allopurinol has been tested as an augmentation strategy for the treatment of mania in bipolar disorder. Meta-analytic evidence showed that adjunctive allopurinol was superior to placebo for acute mania (both with and without mixed features).[17] Its efficacy was not influenced by dosage, follow-up duration, or concurrent standard treatment.[17]
Side effects
Because allopurinol is not a uricosuric, it can be used in people with poor kidney function. However, for people with impaired kidney function, allopurinol has two disadvantages. First, its dosing is complex.[18] Second, some people are hypersensitive to the drug; therefore, its use requires careful monitoring.[19][20]
Allopurinol has rare but potentially fatal adverse effects involving the skin. The most serious adverse effect is a hypersensitivity syndrome consisting of fever, skin rash, eosinophilia, hepatitis, and worsened renal function, collectively referred to as DRESS syndrome.[19] Allopurinol is one of the drugs commonly known to cause Stevens–Johnson syndrome and toxic epidermal necrolysis, two life-threatening dermatological conditions.[19] More common is a less-serious rash that leads to discontinuing this drug.[19]
More rarely, allopurinol can also result in the depression of bone marrow elements, leading to cytopenias, as well as aplastic anemia. Moreover, allopurinol can also cause peripheral neuritis in some patients, although this is a rare side effect. Another side effect of allopurinol is interstitial nephritis.[21]
Allopurinol should not be given to people who are allergic to it.[10]
Drug interactions
Drug interactions are extensive, and are as follows:[10]
Azathioprine and 6-mercaptopurine: Azathioprine is metabolised to 6-mercaptopurine which in turn is inactivated by the action of xanthine oxidase – the target of allopurinol. Giving allopurinol with either of these drugs at their normal dose will lead to overdose of either drug; only one-quarter of the usual dose of 6-mercaptopurine or azathioprine should be given;
Didanosine: plasma didanosine Cmax and AUC values were approximately doubled with concomitant allopurinol treatment; it should not be co-administered with allopuroinol and if it must be, the dose of should be reduced and the person should be closely monitored.
Allopurinol may also increase the activity or half-life of the following drugs, in order of seriousness and certainty of the interaction:[10]
A common misconception is that allopurinol is metabolized by its target, xanthine oxidase, but this action is principally carried out by aldehyde oxidase.[22] The active metabolite of allopurinol is oxipurinol, which is also an inhibitor of xanthine oxidase. Allopurinol is almost completely metabolized to oxipurinol within two hours of oral administration, whereas oxipurinol is slowly excreted by the kidneys over 18–30 hours. For this reason, oxipurinol is believed responsible for the majority of allopurinol’s effect.[23]
Mechanism of action
Allopurinol is a purine analog; it is a structural isomer of hypoxanthine (a naturally occurring purine in the body) and is an inhibitor of the enzyme xanthine oxidase.[2] Xanthine oxidase is responsible for the successive oxidation of hypoxanthine and xanthine, resulting in the production of uric acid, the product of human purine metabolism.[2] In addition to blocking uric acid production, inhibition of xanthine oxidase causes an increase in hypoxanthine and xanthine. While xanthine cannot be converted to purine ribotides, hypoxanthine can be salvaged to the purine ribotidesadenosine and guanosine monophosphates. Increased levels of these ribotides may cause feedback inhibition of amidophosphoribosyl transferase, the first and rate-limiting enzyme of purine biosynthesis. Allopurinol, therefore, decreases uric acid formation and may also inhibit purine synthesis.[24]
Pharmacogenetics
The HLA-B*5801 allele is a genetic marker for allopurinol-induced severe cutaneous adverse reactions, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN).[25][26] The frequency of the HLA-B*5801 allele varies between ethnicities: Han Chinese and Thai populations have HLA-B*5801 allele frequencies of around 8%, as compared to European and Japanese populations, who have allele frequencies of around 1.0% and 0.5%, respectively.[27] The increase in risk for developing allopurinol-induced SJS or TEN in individuals with the HLA-B*5801 allele (as compared to those who do not have this allele) is very high, ranging from a 40-fold to a 580-fold increase in risk, depending on ethnicity.[25][26] As of 2011 the FDA-approved drug label for allopurinol did not contain any information regarding the HLA-B*5801 allele, though FDA scientists did publish a study in 2011 which reported a strong, reproducible and consistent association between the allele and allopurinol-induced SJS and TEN.[28] However, the American College of Rheumatology recommends screening for HLA-B*5801 in high-risk populations (e.g. Koreans with stage 3 or worse chronic kidney disease and those of Han Chinese and Thai descent), and prescribing patients who are positive for the allele an alternative drug.[29] The Clinical Pharmacogenetics Implementation Consortium guidelines state that allopurinol is contraindicated in known carriers of the HLA-B*5801 allele.[30][31]
History
Allopurinol was first synthesized and reported in 1956 by Roland K. Robins (1926-1992), in a search for antineoplastic agents.[2][32] Because allopurinol inhibits the breakdown (catabolism) of the thiopurine drug mercaptopurine, and it was later tested by Wayne Rundles, in collaboration with Gertrude Elion‘s lab at Wellcome Research Laboratories to see if it could improve treatment of acute lymphoblastic leukemia by enhancing the action of mercaptopurine.[2][33] However, no improvement in leukemia response was noted with mercaptopurine-allopurinol co-therapy, so that work turned to other compounds and the team then started testing allopurinol as a potential for gout.[34] Allopurinol was first marketed as a treatment for gout in 1966.[33]
Society and culture
Pure allopurinol is a white powder.
Formulations
Allopurinol is sold as an injection for intravenous use[12] and as a tablet.[10]
Brands
Allopurinol has been marketed in the United States since 19 August 1966, when it was first approved by FDA under the trade name Zyloprim.[35] Allopurinol was marketed at the time by Burroughs-Wellcome. Allopurinol is a generic drug sold under a variety of brand names, including Allohexal, Allosig, Milurit, Alloril, Progout, Ürikoliz, Zyloprim, Zyloric, Zyrik, and Aluron.[36]
^ Jump up to:abcdefg“Allopurinol”. The American Society of Health-System Pharmacists. Archived from the original on 29 April 2016. Retrieved 8 December 2016.
^ Jump up to:ab Robinson PC, Stamp LK (May 2016). “The management of gout: Much has changed”. Australian Family Physician. 45 (5): 299–302. PMID27166465.
^ Satpanich, P; Pongsittisak, W; Manavathongchai, S (18 August 2021). “Early versus Late Allopurinol Initiation in Acute Gout Flare (ELAG): a randomized controlled trial”. Clinical Rheumatology. doi:10.1007/s10067-021-05872-8. PMID34406530. S2CID237156638.
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Sparrow MP, Hande SA, Friedman S, Cao D, Hanauer SB (February 2007). “Effect of allopurinol on clinical outcomes in inflammatory bowel disease nonresponders to azathioprine or 6-mercaptopurine”. Clinical Gastroenterology and Hepatology. 5 (2): 209–14. doi:10.1016/j.cgh.2006.11.020. PMID17296529.
^ Jump up to:ab Bartoli F, Cavaleri D, Bachi B, Moretti F, Riboldi I, Crocamo C, Carrà G (September 2021). “Repurposed drugs as adjunctive treatments for mania and bipolar depression: A meta-review and critical appraisal of meta-analyses of randomized placebo-controlled trials”. Journal of Psychiatric Research. 143: 230–238. doi:10.1016/j.jpsychires.2021.09.018. PMID34509090. S2CID237485915.
^ Reiter S, Simmonds HA, Zöllner N, Braun SL, Knedel M (March 1990). “Demonstration of a combined deficiency of xanthine oxidase and aldehyde oxidase in xanthinuric patients not forming oxipurinol”. Clinica Chimica Acta; International Journal of Clinical Chemistry. 187 (3): 221–34. doi:10.1016/0009-8981(90)90107-4. PMID2323062.
^ Day RO, Graham GG, Hicks M, McLachlan AJ, Stocker SL, Williams KM (2007). “Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol”. Clinical Pharmacokinetics. 46 (8): 623–44. doi:10.2165/00003088-200746080-00001. PMID17655371. S2CID20369375.
^ Cameron JS, Moro F, Simmonds HA (February 1993). “Gout, uric acid and purine metabolism in paediatric nephrology”. Pediatric Nephrology. 7 (1): 105–18. doi:10.1007/BF00861588. PMID8439471. S2CID34815040.
Common side effects include headache, nausea, sleepiness, and unsteadiness.[2] It is unclear if use during pregnancy is safe for the baby.[2] Fomepizole works by blocking the enzyme that converts methanol and ethylene glycol to their toxic breakdown products.[2]
Fomepizole was approved for medical use in the United States in 1997.[2] It is on the World Health Organization’s List of Essential Medicines.[3]FomepizoleCAS Registry Number: 7554-65-6 CAS Name: 4-Methyl-1H-pyrazole Additional Names: 4-MP Trademarks: Antizol (Orphan Med.) Molecular Formula: C4H6N2, Molecular Weight: 82.10 Percent Composition: C 58.52%, H 7.37%, N 34.12% Literature References: Alcohol dehydrogenase inhibitor. Prepn: H. Pechmann, E. Burkard, Ber.33, 3590 (1900); D. S. Noyce et al.,J. Org. Chem.20, 1681 (1955); T. Momose et al.,Heterocycles30, 789 (1990). Inhibition of human liver alcohol dehydrogenase: T.-K. Li, H. Theorell, Acta Chem. Scand.23, 892 (1969). Toxicity study: G. Magnusson et al.,Experientia28, 1198 (1972). GC determn in plasma and urine: R. Achari, M. Mayersohn, J. Pharm. Sci.73, 690 (1984). Clinical pharmacology: D. Jacobsen et al.,Alcohol. Clin. Exp. Res.12, 516 (1988). Pharmacokinetics: eidem,Eur. J. Clin. Pharmacol.37, 599 (1989). Clinical trial in ethylene glycol poisoning: J. Brent et al.,N. Engl. J. Med.340, 832 (1999); in methanol poisoning: idem et al., ibid.344, 424 (2001). Review: J. Likforman et al.,J. Toxicol. Clin. Exp.7, 373-382 (1987). Review of use in methanol poisoning: M. B. Mycyk, J. B. Leikin, Am. J. Therapeut.10, 68-70 (2003). Properties: mp 15.5-18.5°. bp18mm 98.5-99.5°; bp730 204-205°. nD22 1.4913. uv max in 95% ethanol: 220 nm (log e 3.47); in 6N HCl: 226 nm (log e 3.65). Sol in water, alcohol. LD50 (7 days) in mice, rats (mmol/kg): 3.8, 3.8 i.v.; 7.8, 6.5 orally (Magnusson). Melting point: mp 15.5-18.5° Boiling point: bp18mm 98.5-99.5°; bp730 204-205° Index of refraction:nD22 1.4913 Absorption maximum: uv max in 95% ethanol: 220 nm (log e 3.47); in 6N HCl: 226 nm (log e 3.65) Toxicity data: LD50 (7 days) in mice, rats (mmol/kg): 3.8, 3.8 i.v.; 7.8, 6.5 orally (Magnusson) Therap-Cat: Antidote to methanol and ethylene glycol poisoning. Therap-Cat-Vet: Antidote to ethylene glycol poisoning in dogs. Keywords: Antidote (Methanol and Ethylene Glycol Poisoning).
Fomepizole was approved by the U.S. Food and Drug Administration (FDA) on Dec 4, 1997. It was developed and marketed as Antizol® by Paladin in the US.
Fomepizole is a competitive alcohol dehydrogenase inhibitor, Alcohol dehydrogenase catalyzes the oxidation of ethanol to acetaldehyde, and it also catalyzes the initial steps in the metabolism of ethylene glycol and methanol to their toxic metabolites. Antizol® is indicated as an antidote for ethylene glycol (such as antifreeze) or methanol poisoning, or for use in suspected ethylene glycol or methanol ingestion, either alone or in combination with hemodialysis.
Antizol® is available as injection solution for intravenous use, containing 1 g/ml of free Fomepizole. The recommended dose is 15 mg/kg should be administered, followed by doses of 10 mg/kg every 12 hours for 4 doses, then 15 mg/kg every 12 hours thereafter until ethylene glycol or methanol concentrations are undetectable or have been reduced below 20 mg/dL.
Approval Date
Approval Type
Trade Name
Indication
Dosage Form
Strength
Company
Review Classification
1997-12-04
First approval
Antizol
Methanol or ethylene glycol poisoning
Injection
1 g/mL
Paladin
Orphan
SYN
SYN
CAS-RN
Formula
Chemical Name
CAS Index Name
5920-30-9
C4H8N2
4,5-dihydro-4-methylpyrazole
7803-57-8
H6N2O
hydrazine hydrate
Hydrazine, monohydrate
78-85-3
C4H6O
methacrolein
2-propenal, 2-methyl-
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
SYN
Reference:
US7553863B2.
https://patents.google.com/patent/US7553863B2/enEthylene glycol is commonly available as automobile radiator antifreeze. Because of its sweet taste, improperly stored antifreeze is a common source of ethylene glycol poisoning, particularly in children. Ethylene glycol is rapidly absorbed from the gastrointestinal tract. Toxicity can be divided into three stages:
4-Methylpyrazole, marketed as Antizol® (fomepizole) by Orphan Medical, Inc. is a specific antidote for the treatment of ethylene glycol poisoning. It works by inhibiting the enzyme alcohol dehydrogenase which is responsible for the conversion of ethylene glycol, which itself is relatively non-toxic, into its toxic metabolites that in turn cause the renal injury and metabolic acidosis. Antizol® is currently approved by the FDA as an antidote for ethylene glycol poisoning or suspected ethylene glycol poisoning and is recommended by poison control centers as first line therapy. See Antizol® (fomepizole) Injection, Product Monograph, Orphan Medical, Inc., 2001, the entire contents of which are hereby incorporated by reference.Methanol is commonly available in the home in automobile windshield washer fluid and as a gas line anti-icing additive. Methanol has a minor degree of direct toxicity. Its major toxicity follows its metabolism to formic acid. Antizol® is also a specific antidote for the treatment of methanol toxicity. It works by inhibiting the enzyme alcohol dehydrogenase which is responsible for the conversion of methanol into its toxic metabolites, formaldehyde and formic acid. Again, Antizol® is approved by the FDA for use in treating methanol poisoning or suspected methanol poisoning and is recommended by poison control centers as first line therapy.Known methods of preparing 4-methylpyrazole include the reaction of alpha, beta-unsaturated carbonyl compounds or diketones with hydrazine or hydrazine derivatives or the dehydrogenation of the corresponding 2-pyrazoline. See U.S. Pat. Nos. 3,200,128, 4,996,327, and 5,569,769. Other processes for preparing 4-methylpyrazole are disclosed in U.S. Pat. Nos. 6,229,022, 5,569,769, and 4,996,327.4-methylpyrazole prepared by synthetic routes employed heretofore may contain impurities and toxic by-products, including pyrazole, hydrazine, and nitrobenzaldehyde. Pyrazole, like 4-methylpyrazole, is also an inhibitor of alcohol dehydrogenase, but is more toxic than 4-methylpyrazole. Pyrazole is a known teratogen (Eisses, 1995) with 10 fold less potency against alcohol dehydrogenase (T. Li et al., Acta Chem. Scan. 1969, 23, 892-902). In addition, Ewen MacDonald published a paper in 1976 that showed pyrazole in contrast to 4-methylpyrazole has a detrimental effect on brain levels of noradrenaline (E. MacDonald, Acta Pharmacol. et Toxicol. 1976, 39, 513-524). Hydrazine and nitrobenzaldehyde are known mutagens and carcinogens (H. Kohno et al., Cancer Sci. 2005, 96, 69-76).These impurities and toxic by-products have been tolerated heretofore because methods of making ultrapure 4-methylpyrazole have not been available. The FDA has previously approved up to 0.5% pyrazole in Antizol®, but recently is requesting a higher level of purity of less than 0.1% pyrazole to qualify such high levels with animal and other studies. Therefore, while the purity of Antizol® is sufficiently high for its antidotal use in emergency medicine, such toxic impurities are not ideal. For example a pregnant woman who needs antidote therapy would risk exposure of a fetus to potentially toxic pyrazole of known teratogenicity and potentially high levels of known carcinogens. Therefore, a need exists for a 4-methylpyrzaole with even lower amounts of pyrazole and other impurities and for a synthesis of such an ultrapure 4-methylpyrazole.The process of the present invention is set forth in the following exemplary scheme:
EXAMPLE 1Preparation of 1,1-diethoxypropane 1Into a 2-liter flask under nitrogen were added 586 g (3.96 moles) of triethyl orthoformate, 46 g (56 ml, 1 mole) of ethanol, and 16 g of ammonium nitrate. Over the course of one hour 232 g (4 moles) of propionaldehyde were added with stirring. An ice bath was used as necessary to keep maintain the mixture at 30-36° C. The mixture turned yellow orange after one-third of the propionaldehyde had been added. The mixture was stirred overnight at room temperature and then brought to pH 7.5±0.2 with 10% aqueous sodium carbonate (about 30 ml). The aqueous layer was decanted, and the organic layer was distilled over sodium carbonate at atmospheric pressure to produce 124 g (81.6%) of 1.
EXAMPLE 2Preparation of 1-ethoxy-1-propene 2Into a 500 ml flask equipped with a 12″×¾″ packed column were added 0.25 g (0.0013 moles) of p-toluene sulfonic acid, followed by 241 g (1.82 moles) of 1. Nitrogen was bubbled into the mixture while 0.157 g (0.00065 moles) of bis(2-ethylhexyl)amine were added. The nitrogen flow was reduced, and the mixture was distilled to 160° C. to partially remove ethyl alcohol and 1-ethoxy-1-propene. The reaction mixture washed with 320 ml of water and then with 70 ml of water. The organic layer was dried over magnesium sulfate and filtered to produce 121 g (77.5%) of 2, bp 67-76° C., as a clear, colorless liquid. Gas chromatographic analysis showed less than 0.01% ethylvinyl ether.
EXAMPLE 3Preparation of 1,1,3,3-tetraethoxy-2-methylpropane 3Into a 5 liter flask equipped with a mechanical stirrer were added 790 g (5.34 moles) of triethyl orthoformate and 4.28 ml of boron trifluoride-diethyl etherate under a nitrogen atmosphere. Temperature was maintained at 25° C. with cooling as needed. To this mixture were added 230 g (2.67 moles) of 1-ethoxy-1-propene were added slowly and dropwise. The reaction mixture was exothermic; the temperature rose to about 35-38° C. The pot was cooled to 25° C. and stirring was continued for one hour. Solid anhydrous sodium carbonate (32.1 g, 0.3 moles) was added in one portion to the flask and stirring was continued for one hour. The mixture was filtered and the filtrate was fractionally distilled under reduced pressure. The light fraction was removed at a pot temperature of 55-60° C. at 10 mm pressure. The vacuum was improved to 3 mm and the pot temperature was permitted to rise to about 100-140° C. to produce 500 g (80%) of 3, bp 80-81° C. at 3 mm, as a clear, colorless to yellow-brown liquid.
EXAMPLE 4Preparation of 4-methylpyrazoleInto a 5 liter flask equipped with a mechanical stirrer were added 1750 ml of sterile USP water to which 266.7 g (2.05 moles) of hydrazine hydrosulfate were added gradually over one hour with stirring. To the above mixture was added dropwise 481 g (2.053 moles) of 3 and the reaction mixture was warmed to 80° C. Heating and stirring were maintained for 3 hours, the flask was cooled to 40° C., and the volatile components were distilled off under a reduced pressure of about 125 mm. The resulting mixture was cooled to 10° C. first with water and then with glycol; 20 ml of water were added to the flask, and cooling was continued to a temperature of 3° C. Thereafter 50% sodium hydroxide solution was added with cooling so as to maintain the temperature below 30° C. The pH of the reaction mixture should be between 4 and 6. A solution of sodium bicarbonate containing 4.9 g of sodium bicarbonate to 55 ml of water was added to the flask. Additional sodium bicarbonate solution was added until the pH reached 7.0. The flask temperature was allowed to rise to 27° C. with continued stirring. The contents of the flask were extracted with ethyl acetate and the aqueous layer was separated. The organic layer was dried over magnesium sulfate, filtered, and the extract was distilled under vacuum. The light fraction was removed at a pot temperature of 55-60° C. at 125 mm pressure. The vacuum was improved to 5 mm for the remainder of the distillation; pot temperatures were permitted to rise to 100-110° C. to produce 134.8 g (84% based on 3) of 4-methylpyrazole, bp 77-80° C. at 5 mm, as a clear, colorless to yellow liquid. Gas chromatographic analysis showed less than 0.1% pyrazole and less than 10 ppm hydrazine.
SYN
Syn
Journal of the American Chemical Society (1949), 71, 3994-4000.
Fomepizole is an alcohol dehydrogenase inhibitor originally commercialized in 1998 by Orphan Medical as an antidote for ethylene glycol (such as antifreeze) or methanol poisoning, or for use in suspected ethylene glycol or methanol ingestion, either alone or in combination with hemodialysis. In January 2015, Takeda launched the product for the treatment of ethylene glycol and methanol poisoning in Japan. Raptor Pharmaceuticals (currently Horizon Therapeutics) was evaluating the compound in phase II clinical studies for the treatment of the symptoms associated with alcohol intolerance due to ALDH2 deficiency; however, no recent developments have been reported. The compound has been licensed to Paladin and Swedish Orphan Biovitrum (formerly Swedish Orphan). Prior to being acquired by Alliance Pharma in 2010, Cambridge Laboratories obtained a license to fomepizole. In 2005, Orphan Medical was acquired by Jazz Pharmaceuticals. In 2011, Takeda licensed the product from Paladin for development and commercialization rights in Japan. In 2015, orphan drug designation in Australia was assigned to the compound for the treatment of ethylene glycol and methanol poisonings. In 2015, the product was acquired by EUSA Pharma from Jazz Pharmaceuticals for the treatment of poisoning. In 2021, the compound was granted orphan drug designation in the U.S. for the treatment of acetaminophen overdose.
Fomepizole is used to treat ethylene glycol and methanol poisoning. It acts to inhibit the breakdown of these toxins into their active toxic metabolites. Fomepizole is a competitive inhibitor of the enzyme alcohol dehydrogenase,[4] found in the liver. This enzyme plays a key role in the metabolism of ethylene glycol, and of methanol.
Ethylene glycol is first metabolized to glycolaldehyde by alcohol dehydrogenase. Glycolaldehyde then undergoes further oxidation to glycolate, glyoxylate, and oxalate. Glycolate and oxalate are the primary toxins responsible for the metabolic acidosis, and for the renal damage, seen in ethylene glycol poisoning.
Methanol is first metabolized to formaldehyde by alcohol dehydrogenase. Formaldehyde then undergoes further oxidation, via formaldehyde dehydrogenase, to become formic acid.[5] Formic acid is the primary toxin responsible for the metabolic acidosis, and for the visual disturbances, associated with methanol poisoning.
By competitively inhibiting the first enzyme, alcohol dehydrogenase, in the metabolism of ethylene glycol and methanol, fomepizole slows the production of the toxic metabolites. The slower rate of metabolite production allows the liver to process and excrete the metabolites as they are produced, limiting the accumulation in tissues such as the kidney and eye. As a result, much of the organ damage is avoided.[6]
Fomepizole is most effective when given soon after ingestion of ethylene glycol or methanol. Delaying its administration allows for the generation of harmful metabolites.[6]
Interaction with alcohol
Concurrent use with ethanol is contraindicated because fomepizole is known to prolong the half-life of ethanol via inhibiting its metabolism. Extending the half-life of ethanol may increase and extend the intoxicating effects of ethanol, allowing for greater (potentially dangerous) levels of intoxication at lower doses. Fomepizole slows the production of acetaldehyde by inhibiting alcohol dehydrogenase, which in turn allows more time to further convert acetaldehyde into acetic acid by acetaldehyde dehydrogenase. The result is a patient with a prolonged and deeper level of intoxication for any given dose of ethanol, and reduced “hangover” symptoms (since these adverse symptoms are largely mediated by acetaldehyde build up).
In a chronic alcoholic who has built up a tolerance to ethanol, this removes some of the disincentives to ethanol consumption (“negative reinforcement“) while allowing them to become intoxicated with a lower dose of ethanol. The danger is that the alcoholic will then overdose on ethanol (possibly fatally). If alcoholics instead very carefully reduce their doses to reflect the now slower metabolism, they may get the “rewarding” stimulus of intoxication at lower doses with less adverse “hangover” effects – leading potentially to increased psychological dependency. However, these lower doses may therefore produce less chronic toxicity and provide a harm minimization approach to chronic alcoholism.
It is, in essence, the antithesis of a disulfiram approach which tries to increase the buildup of acetaldehyde resulting in positive punishment for the patient. Compliance, and adherence, is a substantial problem in disulfiram-based approaches. Disulfiram also has a considerably longer half-life than that of fomepizole, requiring the person to not drink ethanol in order to avoid severe effects. If the person is not adequately managed on a benzodiazepine, barbiturate, acamprosate, or another GABAA receptor agonist, the alcohol withdrawal syndrome, and its attendant, life-threatening risk of delirium tremens “DT”, may occur. Disulfiram treatment should never be initiated until the risk of DT has been evaluated, and mitigated appropriately. Fomepizole treatment may be initiated while the DT de-titration sequence is still being calibrated based upon the person’s withdrawal symptoms and psychological health.[citation needed]
Adverse effects
Common side effects associated with fomepizole use include headache and nausea.[7]
Kinetics
Absorption and distribution
Fomepizole distributes rapidly into total body water. The volume of distribution is between 0.6 and 1.02 L/kg. The therapeutic concentration is from 8.2 to 24.6 mg (100 to 300 micromoles) per liter. Peak concentration following single oral doses of 7 to 50 mg/kg of body weight occurred in 1 to 2 hours. The half-life varies with dose, so has not been calculated.
Metabolism and elimination
Hepatic; the primary metabolite is 4-carboxypyrazole (about 80 to 85% of an administered dose). Other metabolites include the pyrazoles 4-hydroxymethylpyrazole and the N -glucuronide conjugates of 4-carboxypyrazole and 4-hydroxymethylpyrazole.
Following multiple doses, fomepizole rapidly induces its own metabolism via the cytochrome P450 mixed-function oxidase system.
In healthy volunteers, 1.0 to 3.5% of an administered dose was excreted unchanged in the urine. The metabolites also are excreted unchanged in the urine.
^ Jump up to:abcdefg“Fomepizole”. The American Society of Health-System Pharmacists. Archived from the original on 21 December 2016. Retrieved 8 December 2016.
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Lepik, KJ; Levy, AR; Sobolev, BG; Purssell, RA; DeWitt, CR; Erhardt, GD; Kennedy, JR; Daws, DE; Brignall, JL (April 2009). “Adverse drug events associated with the antidotes for methanol and ethylene glycol poisoning: a comparison of ethanol and fomepizole”. Annals of Emergency Medicine. 53 (4): 439–450.e10. doi:10.1016/j.annemergmed.2008.05.008. PMID18639955.
^ Vos, Johannes G.; Groeneveld, Willem L. (1979). “Pyrazolato and related anions. Part V. Transition metal salts of 4-methylpyrazole”. Transition Metal Chemistry. 4 (3): 137–141. doi:10.1007/BF00619054. S2CID93580021.
External links
“Fomepizole”. Drug Information Portal. U.S. National Library of Medicine.
(2S)-2-(3-aminopropanamido)-3-(1H-imidazol-5-yl)propanoic acid (E)-N-(3-Amino-1-hydroxypropylidene)-L-histidine [ACD/IUPAC Name] 206-169-9[EINECS], 305-84-0[RN] 8HO6PVN24Wカルノシン , Dragosine, Ignotin, Ignotine, Karnozin, L-Carnosine, N-(β-Alanyl)-L-histidine, NSC 524045, Sevitin, β-Alanylhistidine CarnosineCAS Registry Number: 305-84-0CAS Name: b-Alanyl-L-histidine Additional Names: ignotine Molecular Formula: C9H14N4O3, Molecular Weight: 226.23 Percent Composition: C 47.78%, H 6.24%, N 24.77%, O 21.22% Literature References: Naturally occurring dipeptide found in large amounts in skeletal muscle. Also present in other tissues such as brain, cardiac muscle, kidney. Water soluble antioxidant; functions as a free-radical scavenger. Isoln: Gulewitsch, Amiradzibi, Ber.33, 1902 (1900); Wolff, Wilson, J. Biol. Chem.95, 495 (1932); 109, 565 (1935). Synthesis from histidine and b-iodo- or b-nitropropionyl chloride: Baumann, Ingvaldsen, ibid.35, 271 (1918); Barger, Tutin, Biochem. J.12, 406 (1918). Later syntheses: Sifford, du Vigneaud, J. Biol. Chem.108, 753 (1935); R. A. Turner, J. Am. Chem. Soc.75, 2388 (1953); F. J. Vinick, S. Jung, J. Org. Chem.48, 392 (1983). Crystal structure: H. Itoh et al.,Acta Crystallogr.33B, 2959 (1977). Possible role in wound healing: D. E. Fischer et al.,Proc. Soc. Exp. Biol. Med.158, 402 (1978). Review of physiological properties and therapeutic potential: S. E. Gariballa, A. J. Sinclair, Age Ageing29, 207-210 (2000). Properties: Crystals from aqueous ethanol, mp 262° (dec) (Vinick, Jung); also reported as mp 260° (capillary tube) and as mp 308-309° (Dennis bar) (Sifford, du Vigneaud). [a]D25 +21.0° (c = 1.5 in water). pK1 2.64; pK2 6.83; pK3 9.51. Alkaline reaction. One gram dissolves in 3.1 ml water at 25°. Melting point: mp 262° (dec) (Vinick, Jung); mp 260° (capillary tube) and as mp 308-309° (Dennis bar) (Sifford, du Vigneaud) pKa: pK1 2.64; pK2 6.83; pK3 9.51 Optical Rotation: [a]D25 +21.0° (c = 1.5 in water) Derivative Type: Nitrate CAS Registry Number: 5852-98-2 Molecular Formula: C9H15N5O6, Molecular Weight: 289.25 Percent Composition: C 37.37%, H 5.23%, N 24.21%, O 33.19% Properties: Crystals, dec 222°. [a]D20 +24.1° (c = 1.5 in water). Very sol in water. Optical Rotation: [a]D20 +24.1° (c = 1.5 in water) Derivative Type: Hydrochloride CAS Registry Number: 5852-99-3 Molecular Formula: C9H15ClN4O3, Molecular Weight: 262.69 Percent Composition: C 41.15%, H 5.76%, Cl 13.50%, N 21.33%, O 18.27% Properties: Crystals, dec 245°. Very sol in water. Derivative Type: D-Form CAS Registry Number: 5853-00-9 Properties: Crystals, mp 260°. [a]D28 -20.4° (c = 1.5). Melting point: mp 260° Optical Rotation: [a]D28 -20.4° (c = 1.5)
Carnosine is naturally produced by the body in the liver[3] from beta-alanine and histidine. Like carnitine, carnosine is composed of the root word carn, meaning “flesh”, alluding to its prevalence in meat.[4] There are no plant-based sources of carnosine,[5] however synthetic supplements do exist.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Example 1 (S) -2- (Cyanoacetylamino) -3- (l_ * H-imidazol-4-yl) propionic acid, sodium salt
To a solution of sodium ethoxide obtained by dissolving 5.57 g (0.24 mol) of sodium in 800 ml of ethanol was added 40.0 g (0.26 mol) of L-histidine at room temperature. After 15 minutes, 44.12 g (0.39 mol) of ethyl cyanoacetate were added and the suspension was refluxed for 16 hours. After cooling to room temperature, the mixture was filtered. The yellowish filtrate was concentrated in vacuo, the residue was slurried in ethyl acetate, filtered, washed with ethyl acetate and purified by flash chromatography on silica gel (eluent: gradient ethyl acetate → methanol / ethyl acetate 3: 1). Yield: 28.42 g (46%) 1HNMR (DMSO- ^ 6, 00 MHz): δ = 8,28 (d, 1H); 7,45 (s, 1H); 6,7 (s, 1H); 5,5 (br. s, 1H); 4,12-4,20 (m, 1H); 3,65 (s, 2H); 2,95-3,05 (m, 1H); 2,8-2,9 (m, 1H). 13C NMR (DMSO- 6, 100 MHz): δ = 174,05; 161,09; 134,25; 131,97; 119,66; 116,43; 54,83; 29,13; 25,20.
Example 2 (• S) -2- (Cyanoacetylamino) -3- (1-δ-imidazol-4-yl) propionic acid, sodium salt
9.80 g of sodium hydride (60% in mineral oil) and 50.6 g (0.51 mol) were added at room temperature to a suspension of 40.0 g (0.26 mol) of L-histidine in 750 ml of N, N-dimethylformamide Given methyl cyanoacetate. The mixture was heated to 155 ° C. for 2 h in an open flask and the solution thus obtained was analyzed by means of HPLC. Histidine (8 area%) and (S) -2- (cyanoacetylamino) -3- (1H-imidazol-4-yl) propionic acid sodium salt (38 area%) were identified.
Example 3 (S) -2- (Cyanoacetylamino) -3- (l-ö r -imidazol-4-yl) propionic acid
To a solution of sodium ethoxide obtained by dissolving 4.02 g (0.175 mol) of sodium in 280 ml of ethanol, 28.27 g (0.18 mol) of L-Ηistidine were added at room temperature. The mixture was heated slowly and 30.92 g (0.27 mol) of ethyl cyanoacetate were added dropwise at a temperature of 60.degree. The mixture was heated further and the ethanol was distilled off, the amount of ethanol distilled off being continuously replaced in portions by N, N-dimethylformamide. At the end of the reaction, the temperature of the solution was 130 ° C. The mixture was stirred at this temperature for a further 2 hours. The brown reaction mixture (200 g) was cooled to 50 ° C. and 30 g of concentrated hydrochloric acid were metered in. About 70 g of solvent (Η 2O / N, N-dimethylformamide mixture) distilled off. The viscous suspension was mixed with 200 g of acetone, cooled to -10 ° C. and filtered. For recrystallization, the residue was dissolved in water and the pH was adjusted to 5.0. On cooling (<5 ° C.) a white solid precipitated out, which was filtered off, washed with ethanol and dried at 40 ° C./20 mbar. Yield: 26.39 g (66%). IR (KBr): v = 3421, 3240, 3149, 3059, 2970, 2255, 1653, 1551, 1396, 1107, 1088, 979, 965, 826, 786, 638 cm is “1 . 1HΝMR (DMSO-c 6 , 400 MHz): δ = 11.0 (br., 2H); 8.50 (d, 1H); 7.68 (s, 1H); 6.85 (s, 1H); 4.35-4.48 ( m, 1H); 3.68 (s, 2H); 2.92-3.03 (, 1H); 2.82-2.91 (m, 1H).
13 C NMR (DMSO- 6 , 100 MHz): δ = 172.23; 161.92; 134.55; 132.70; 116.73; 115.87; 52.80; 28.68; 25.06. LC-MS: mlz = 223 ([M + H]), 205, 177, 156, 110. The optical purity was determined to be> 99.8% on a sample obtained according to the above procedure. The determination was carried out by hydrolysis of the amide bond (6 N hydrochloric acid, 110 ° C., 24 h), followed by derivatization of the released histidine with trifluoroacetic anhydride and isobutyl chloroformate. A D-histidine content of <0.1% was detected by gas chromatography on a chiral stationary phase.
Example 4 L-Carnosine
To a solution of 1.90 g (7.8 mmol) of (<S) -2- (cyanoacetylamino) -3- (1H-imidazol-4-yl) propionic acid sodium salt (prepared according to Example 1) in 50 ml of ethanol / conc. Ammonia solution (V: V- 4: 1) were given 0.3 g of rhodium / activated charcoal (5% Rh). The
The mixture was hydrogenated at 110 ° C. and 45 bar for 1 hour. The catalyst was then filtered off and the filtrate was adjusted to pH 8.2 with formic acid. After the solution had been concentrated in vacuo, the residue was suspended in 200 ml of ethanol and heated to 60 ° C. for 30 minutes. The product was filtered off, washed successively with ethanol, ethyl acetate and diethyl ether and finally dried. Yield: 1.33 g (76%) 1H NMR (D 2 O, 400 MΗz): δ = 7.70 (s, 1Η); 6.93 (s, 1Η); 4.43-4.50 (m, 1Η); 3.20-3.28 (m, 2Η); 3.11-3.19 (m, 1H); 2.95-3.03 (m, 1H); 2.61-2.71 (m, 2H). The optical purity was determined by the method described in Example 3 to be 99.5%.
Example 5 (S) -2- (Cyanoacetylamino) -3- (1-O-imidazol-4-yl) propionic acid methyl ester
To a solution of sodium methoxide obtained by dissolving 0.94 g (40.7 mmol; 1.95 equiv.) Of sodium in 100 ml of methanol, 5.0 g (20.4 mmol) were added at room temperature
L-histidine methyl ester dihydrochloride added. After 30 minutes, 3.03 g (30.6 mmol) of methyl cyanoacetate were added and the mixture was left on for 16 hours
Boiled under reflux. After cooling to room temperature, the mixture was filtered.
The yellowish filtrate was concentrated in vacuo and the residue was purified by means of flash chromatography on silica gel (eluent: gradient ethyl acetate – »ethyl acetate / methanol 3: 1). Yield: 1.51 g (31%) 1H MR (OMSO-de, 400 MHz): δ = 8.65 (d, 1H); 7.52 (s. 1H); 6.8 (s, 1H); 4.45 ^ 1.55 (m,
1.76 g of Rh / C (0.4 mol% of pure Rh based on the starting material used) in a mixture of 94.2 g of ammonia solution (25% in H 2 O) and 62.8 g of methanol were placed in a 1 liter pressure autoclave . The autoclave was closed, the contents were heated to 90 ° C. and 40 bar hydrogen was injected. A solution of 20.0 g (0.09 mol) (* S) -2- (cyanoacetylamino) -3- (1H-imidazol-4-yl) propionic acid (prepared according to Example 3) was then within one hour Mixture 94.2 g ammonia solution (25% in Η 2O) and 62.8 g of methanol are metered in. After a one hour post-reaction at 90 ° C., the reaction mixture was cooled to room temperature. The pressure in the autoclave was released and the catalyst was filtered off over activated charcoal. An HPLC in-process analysis showed that the clear greenish reaction solution (326.2 g) contained 5.74% (m / m) carnosine, which corresponds to a selectivity of 92% with complete conversion. The reaction mixture was then concentrated to approx. 60 g on a rotary evaporator. As a result of the dropwise addition of 174 g of ethanol, a white solid precipitated out, which was filtered off and dried at 50 ° C./20 mbar.
In a 1 liter pressure autoclave, a solution of 10.00 g (45.0 mmol) (S) -2- (cyanoacetylamino) -3 was added to 0.88 g of Rh / C (0.4 mol% of pure Rh based on the starting material used) – (1H-imidazol-4-yl) propionic acid (prepared according to Example 3) in a mixture of 157 g conc. NΗ 3/ Methanol (m / m = 3: 2) was added. The autoclave was closed and flushed twice with 40 bar nitrogen and once with hydrogen. The mixture was heated to 90 ° C. and 40 bar hydrogen was injected. After 3 h at 90 ° C., the reaction mixture was cooled to room temperature, the autoclave was depressurized and the catalyst was separated off by filtration. An in-process analysis (HPLC) showed that the reaction solution (147.2 g) contained 6.38% (m / m) carnosine, which corresponds to a selectivity of 92% when the conversion is complete. The reaction mixture was then concentrated to 41.2 g on a rotary evaporator. 124 g of ethanol were added dropwise at room temperature and the flask was placed in a refrigerator overnight. The next day the precipitate was filtered off, washed with ethanol and dried in a drying cabinet at 40 ° C./20 mbar. 7.96 g (78%) of a slightly greenish solid with a content (HPLC) of 98.0% (m / m) were obtained.
Example 8 L-Carnosine
The procedure was as described in Example 7, with the difference that 5% Rh on aluminum oxide was used as the catalyst. Under these conditions, L-carnosine was formed with 83% selectivity.
Example 9 L-Carnosine
4.5 g of Raney cobalt (doped with 0.3% iron) in 195 g of methanol were placed in a 1 liter pressure autoclave. A solution of 30.0 g (0.135 mol) (S) -2- (cyanoacetylamino) -3- (1H-imidazol-4-yl) propionic acid (prepared according to Example 3) in 375 g ammonia solution (25% in Η O) was admitted. The autoclave was closed and flushed twice with 40 bar nitrogen. Then 45 bar of hydrogen were injected and the contents were heated to 100 ° C. within half an hour. After an after-reaction of 3 hours at 100 ° C., the reaction mixture was cooled to room temperature and the pressure in the autoclave was released. An HPLC in-process analysis showed that the reaction solution (590.8 g) contained 4.68% (mim) carnosine, which corresponds to a selectivity of 91% with complete conversion.
Example 10 L-Carnosine
In a 100 ml pressure autoclave were to a solution of 2.0 g (9.0 mmol) (ιS) -2- (cyanoacetylamino) -3- (lH-imidazol-4-yl) propionic acid (prepared according to Example 3) in a Mixture of 25 g of ammonia solution (25% in Η 2 O) and 13 g of methanol, 1.1 g of Raney nickel (doped with 1.8% molybdenum) were added. The autoclave was closed and placed in an oil bath preheated to 100.degree. After 10 minutes, 50 bar of hydrogen were injected. After 2.5 hours at 100 ° C., the reaction mixture was cooled to room temperature and the pressure on the autoclave was released. An HPLC in-process analysis showed that the reaction solution (39.4 g) contained 4.54% (m / m) carnosine, which, with a conversion of 99%, corresponds to a selectivity of 89%.
Example 11 L-Carnosine
In a 1 liter pressure autoclave, 4.50 g of Raney cobalt (doped with 0.3% iron) in a mixture of 285 g of conc. Ammonia / methanol (mim = 1.9: 1) submitted. The autoclave was closed and flushed twice with 40 bar nitrogen. Then 45 bar of hydrogen were injected and the mixture was heated to 100.degree. A solution of 30.0 g (0.135 mol) of (S) -2- (cyanoacetylamino) -3- (1H-imidazol-4-yl) propionic acid (prepared according to Example 3) in a mixture of 285 g was then obtained within one hour conc. Ammonia / methanol (m / m = 1.9: 1) metered in. After a one hour post-reaction at 100 ° C., the reaction mixture was cooled to room temperature. The pressure in the autoclave was released and the catalyst was filtered off. A ΗPLC in-process analysis showed that the reddish brown reaction solution (310.5 g) contained 9.57% (m / m) carnosine,
Example 12 (S) -2- (Cyanoacetylamino) -3- (3-methyl-3-ö r -imidazol-4-yl) propionic acid, sodium salt
0.50 g (2.95 mmol) of 3-methyl-L-histidine were added at 40 ° C. to a solution of 0.20 g (2.94 mmol) of sodium ethoxide in 5.60 g of ethanol. The clear solution was heated to 60 ° C. and 0.50 g (4.43 mmol) ethyl cyanoacetate was added dropwise. The mixture was refluxed for 1 hour. Then 10 mg (0.15 mmol) of imidazole were added. The ethanol was then slowly distilled off and the amount of ethanol distilled off was continuously replaced in portions by N, N-dimethylformamide. After a subsequent reaction time of 2 h at 125 ° C., the reaction mixture was carefully concentrated and the residue was purified by means of flash column chromatography on silica gel (eluent: gradient ethyl acetate → ethyl acetate / methanol 2: 1). 0.49 g (64%) of a slightly yellowish solid were obtained.
Example 13 (S) -2- (3-aminopropionylamino) -3- (3-methyl-3Jϊ-imidazol-4-yl) propionic acid (= anserine)
To a solution of 0.20 g (0.77 mmol) (5) -2- (cyanoacetylamino) -3- (3-methyl-3H-imidazol-4-yl) propionic acid sodium salt (prepared according to Example 12) in 2 , 4 g of methanol and 1.6 g of ammonia solution (25% in Η 2 O), 16 mg of rhodium / Al 2 O 3 (5% Rh) were added. The mixture was hydrogenated at 85 ° C. and 50 bar for 1 hour. The catalyst was then filtered off. Anserine could be clearly detected in the filtrate by means of thin-layer chromatography, HPLC (by co-injection with a commercial reference substance) and LC-MS. Gross yield: approx. 45%. TLC: R f = 0.25 (ethyl acetate / methanol / Ammom ‘ ak H 2 O 43: 35: 8: 10). LC-MS: m / z = 241 ([M + H] +), 224, 206, 180, 170, 126, 109.
SYN
Synthesis of L-carnosine from two amino acids β -alanine-amide and L-histidine
L-Carnosine (L-Car, β-alanyl-L-histidine) is a bioactive dipeptide with important physiological functions. Direct coupling of unprotected β-Ala (β-alanine) with L-His (L-histidine) mediated by an enzyme is a promising method for L-Car synthesis. In this study, a new recombinant dipeptidase (SmPepD) from Serratia marcescens with a high synthetic activity toward L-Car was identified by a genome mining approach and successfully expressed in Escherichia coli. Divalent metal ions strongly promoted the synthetic activity of SmPepD, with up to 21.7-fold increase of activity in the presence of 0.1 mM MnCl2. Higher temperature, lower pH and increasing substrate loadings facilitated the L-Car synthesis. Pilot biocatalytic syntheses of L-Car were performed comparatively in batch and continuous modes. In the continuous process, an ultra-filtration membrane reactor with a working volume of 5 L was employed for catalyst retention. The dipeptidase, SmPepD, showed excellent operational stability without a significant decrease in space–time yield after 4 days. The specific yield of L-Car achieved was 105 gCar gcatalyst−1 by the continuous process and 30.1 gCar gcatalyst−1 by the batch process. A nanofiltration membrane was used to isolate the desired product L-Car from the reaction mixture by selectively removing the excess substrates, β-Ala and L-His. As a result, the final L-Car content was effectively enriched from 2.3% to above 95%, which gave L-Car in 99% purity after ethanol precipitation with a total yield of 60.2%. The recovered substrate mixture of β-Ala and L-His can be easily reused, which will enable the economically attractive and environmentally benign production of the dipeptide L-Car.
Carnosine is a dipeptide of the amino acids beta-alanine and histidine. It is highly concentrated in muscle and brain tissues.
[0005] β-Alanine (or beta-alanine) is a naturally occurring beta amino acid, which is an amino acid in which the amino group is at the β-position from the carboxylate group (i.e., two atoms away).
[0006] β-Alanine is not used in the biosynthesis of any major proteins or enzymes. It is formed in vivo by the degradation of dihydrouracil and carnosine. It is a component of the naturally occurring peptides carnosine and anserine and also of pantothenic acid (vitamin B5), which itself is a component of coenzyme A. Under normal conditions, β-alanine is metabolized into acetic acid.
[0007] β-Alanine is the rate-limiting precursor of carnosine, which is to say carnosine levels are limited by the amount of available β-alanine, not histidine. Supplementation with β-alanine has been shown to increase the concentration of carnosine in muscles, decrease fatigue in athletes and increase total muscular work done.
[0008] Carnosine and beta-alanine are popular dietary supplements currently produced using chemical methods. Beta-alanine is also a synthetic precursor to pantothenic acid, the essential vitamin B5. Beta-alanine can also be used as a monomer for the production of a polymeric resin (U.S. Pat. No. 4,082,730).
[0009] Naturally, carnosine is produced exclusively in animals from beta-alanine (via uracil) and histidine. In yeasts and animals, beta-alanine is typically produced by degradation of uracil. Chemically, carnosine can be synthesized from histidine and beta-alanine derivatives. For example, the coupling of an N-(thiocarboxy) anhydride of beta-alanine with histidine has been described (Vinick et al. A simple and efficient synthesis of L-carnosine. J. Org. Chem, 1983, 48(3), pp. 392-393).
[0010] Beta-alanine can be produced synthetically by Michael addition of ammonia to ethyl- or methyl-acrylate. This requires the use of the caustic agent ammonia and high pressures. It is also natively produced in bacteria and yeasts in small quantities. In bacteria, beta-alanine is produced by decarboxylation of aspartate. Lysates of bacteria have been used in biocatalytic production from aspartate (Patent CN104531796A).
[0011] There remains a need in the industry for a safer, more economical system for the production of carnosine and beta-alanine.
[0105] The present disclosure provides methods for the biosynthetic production of beta-alanine and carnosine using engineered microorganisms of the present invention.
[0106] In one embodiment, a method of producing beta-alanine is provided. The method comprises providing a fermentation media comprising a carbon substrate, contacting said media with a recombinant yeast microorganism expressing an engineered beta-alanine biosynthetic pathway wherein said pathway comprises an aspartate to beta-alanine conversion (pathway step a), and culturing the yeast in conditions whereby beta-alanine is produced.
[0107] In another embodiment of the present invention, a method of producing carnosine is provided. The method comprises providing a fermentation media comprising a carbon substrate, contacting said media with a recombinant yeast microorganism expressing an engineered carnosine biosynthetic pathway wherein said pathway comprises (i) an aspartate to beta-alanine conversion (pathway step a) and (ii) a beta-alanine to carnosine conversion (pathway step b), and culturing the yeast in conditions whereby carnosine is produced.
[0108] In another embodiment of the present invention, a method of producing carnosine via biotransformation is provided. The method comprises providing a media comprising a carbon substrate and exogenously added beta-alanine, contacting said media with a recombinant yeast microorganism expressing an engineered carnosine biosynthetic pathway wherein said pathway comprises (i) a beta-alanine to carnosine conversion (pathway step b), and culturing the yeast in conditions whereby carnosine is produced.
[0109] Some embodiments of the present invention comprise yeast strains designated ca1 and ca2 and are derived from S. cerevisiae strain S288C. Each encodes at least 2 foreign genes under inducible Gal promoters. Strain ca1 also contains an additional gene, panM. The specific proteins encoded by each strain and their sequences, source, and accession numbers are provided in Table 1. The genes for these proteins are synthesized with yeast-optimized codon usage, assembled into singular genetic cassettes, and then inserted into the HO locus of S288C under URA2 selection. Strains ca1 and ca2 served as parent strains to derivatives comprising various heterologous genes. Ca2 served as a parent strain for ca7, ca8, ca9, ca10, ca11, ca12, ca14, ca15 in which the carnosine synthase is a different ortholog. Strain ca1 served as the parent strain to strains ca19, ca20, ca21, ca22, ca23, ca24, ca27, and ca28 in which the aspartate decarboxylase is a different ortholog. The specific proteins encoded by each strain and their sequences, source, and accession numbers are provided in Table 2.
[0110] Aspartate, histidine, and the cofactors involved in the carnosine and beta-alanine pathway are universal to all organisms, and thus the host organism could be any genetically tractable organism (plants, animals, bacteria, or fungi). Amongst yeasts, other species such as S. pombe or P. pastoris are plausible alternatives. Within the S. cerevisiae species, other strains more amenable to large-scale productions, such as CENPalpha, may be utilized.
[0111] The Gal promoter used in embodiments of the present invention could be replaced with constitutive promoters, or other chemically-inducible, growth phase-dependent, or stress-induced promoters. Heterologous genes of the present invention may be genomically encoded or alternatively encoded on plasmids or yeast artificial chromosomes (YACs). All genes introduced could be encoded with alternate codon usage without altering the biochemical composition of the system. All enzymes used in embodiments of the present invention have extensive orthologs in the biosphere that could be encoded as alternatives.
[0112] Aspartate, histidine, and the cofactors involved in this pathway are universal to all organisms, and thus the host organism could be any genetically tractable organism (plants, animals, bacteria, or fungi). Among yeast, other species such as S. pombe or P. pastoris are plausible alternatives. Within the S. cerevisiae species, other strains more amenable to large scale productions, such as CENPalpha, may be preferable. The panD gene can replaced with orthologs from other bacteria. Examples include Corynebacterium glutamicum Escherichia coli, Helicobacter pylori, Tribolium castaneum, Pectobacterium carotovorum, Actinoplanes sp. SE50/110, Taoultella ornithinolytica, Methanocaldococcus jannaschii DSM 2661 and Methanocaldococcus bathoardescens. This is shown in Table 2. Carnosine synthase is natively found in mammals, birds, and reptiles. Therefore, the chicken enzyme used in ca1 and ca2 could be replaced by various orthologs. Examples include Gorilla gorilla, Falco perefrinus, Allpiucator mississsippiensis, Ailuoropoda melanoleuca, Ursus maritimus, Python bivittatus, and Orcinus orca. This is shown in Table 2.
Culture Conditions
[0113] The growth medium used to test for production of carnosine by the engineered strains was Teknova SC Minimal Broth with Raffinose supplemented with 1% galactose.
[0114] A variety of purification protocols including solid phase extraction and cation exchange chromatography may be employed to purify the desired products from the culture supernatant or the yeast cell pellet fraction.
Carnosine is synthesized within the body from beta-alanine and histidine. Beta-alanine is a product of pyrimidine catabolism[6] and histidine is an essential amino acid. Since beta-alanine is the limiting substrate, supplementing just beta-alanine effectively increases the intramuscular concentration of carnosine.[7][8]
Physiological effects
pH buffer
Carnosine has a pKa value of 6.83, making it a good buffer for the pH range of animal muscles.[9] Since beta-alanine is not incorporated into proteins, carnosine can be stored at relatively high concentrations (millimolar). Occurring at 17–25 mmol/kg (dry muscle),[10] carnosine (β-alanyl-L-histidine) is an important intramuscular buffer, constituting 10-20% of the total buffering capacity in type I and II muscle fibres.
Anti-oxidant
Carnosine has been proven to scavenge reactive oxygen species (ROS) as well as alpha-beta unsaturated aldehydes formed from peroxidation of cell membrane fatty acids during oxidative stress. It also buffers pH in muscle cells, and acts as a neurotransmitter in the brain. It is also a zwitterion, a neutral molecule with a positive and negative end.[citation needed]
Carnosine is considered as a geroprotector.[15] Carnosine can increase the Hayflick limit in human fibroblasts,[16] as well as appearing to reduce the telomere shortening rate.[17] Carnosine may also slow aging through its anti-glycating properties (chronic glycolysis is speculated to accelerate aging).[18]
Carnosine administration has been shown to have cardioprotective properties, protecting against ischaemia-reperfusion injury, and doxorubicin-induced cardiomyopathy.[19]
Carnosine demonstrated neuroprotective effects in multiple animal studies.[20][21][22]
Research has demonstrated a positive association between muscle tissue carnosine concentration and exercise performance.[23][24][25] β-Alanine supplementation is thought to increase exercise performance by promoting carnosine production in muscle. Exercise has conversely been found to increase muscle carnosine concentrations, and muscle carnosine content is higher in athletes engaging in anaerobic exercise.[23]
Carnosine appears to protect in experimental ischemic stroke by influencing a number of mechanisms that are activated during stroke. It is a potent pH buffer and has anti matrix metalloproteinase activity, antioxidant and antiexcitotoxic properties and protects the blood brain barrier [26], [27], [28], [29], [30], [31], [32]. [33], [34], [35].
^ Alan R. Hipkiss (2009). “Chapter 3: Carnosine and Its Possible Roles in Nutrition and Health”. Advances in Food and Nutrition Research.
^“beta-ureidopropionate + H2O => beta-alanine + NH4+ + CO2”. reactome. Retrieved 2020-02-08. Cytosolic 3-ureidopropionase catalyzes the reaction of 3-ureidopropionate and water to form beta-alanine, CO2, and NH3 (van Kuilenberg et al. 2004).
^ Derave W, Ozdemir MS, Harris R, Pottier A, Reyngoudt H, Koppo K, Wise JA, Achten E (August 9, 2007). “Beta-alanine supplementation augments muscle carnosine content and attenuates fatigue during repeated isokinetic contraction bouts in trained sprinters”. J Appl Physiol. 103 (5): 1736–43. doi:10.1152/japplphysiol.00397.2007. PMID17690198. S2CID6990201.
^ Hill CA, Harris RC, Kim HJ, Harris BD, Sale C, Boobis LH, Kim CK, Wise JA (2007). “Influence of beta-alanine supplementation on skeletal muscle carnosine concentrations and high intensity cycling capacity”. Amino Acids. 32 (2): 225–33. doi:10.1007/s00726-006-0364-4. PMID16868650. S2CID23988054.
^ Jump up to:ab Reddy, V. P.; Garrett, MR; Perry, G; Smith, MA (2005). “Carnosine: A Versatile Antioxidant and Antiglycating Agent”. Science of Aging Knowledge Environment. 2005 (18): pe12. doi:10.1126/sageke.2005.18.pe12. PMID15872311.
^ Rashid, Imran; Van Reyk, David M.; Davies, Michael J. (2007). “Carnosine and its constituents inhibit glycation of low-density lipoproteins that promotes foam cell formation in vitro”. FEBS Letters. 581 (5): 1067–70. doi:10.1016/j.febslet.2007.01.082. PMID17316626. S2CID46535145.
^ Boldyrev, A. A.; Stvolinsky, S. L.; Fedorova, T. N.; Suslina, Z. A. (2010). “Carnosine as a natural antioxidant and geroprotector: From molecular mechanisms to clinical trials”. Rejuvenation Research. 13 (2–3): 156–8. doi:10.1089/rej.2009.0923. PMID20017611.
^ McFarland, G; Holliday, R (1994). “Retardation of the Senescence of Cultured Human Diploid Fibroblasts by Carnosine”. Experimental Cell Research. 212 (2): 167–75. doi:10.1006/excr.1994.1132. PMID8187813.
^ Shao, Lan; Li, Qing-Huan; Tan, Zheng (2004). “L-Carnosine reduces telomere damage and shortening rate in cultured normal fibroblasts”. Biochemical and Biophysical Research Communications. 324 (2): 931–6. doi:10.1016/j.bbrc.2004.09.136. PMID15474517.
26. Kim EH, Kim ES, Shin D, Kim D, Choi S, Shin YJ, Kim KA, Noh D, Caglayan AB, Rajanikant GK, Majid A, Bae ON. Carnosine Protects against Cerebral Ischemic Injury by Inhibiting Matrix-Metalloproteinases. Int J Mol Sci. 2021 Jul 13;22(14):7495. doi: 10.3390/ijms22147495. PMID: 34299128; PMCID: PMC8306548.
27. Jain S, Kim ES, Kim D, Burrows D, De Felice M, Kim M, Baek SH, Ali A, Redgrave J, Doeppner TR, Gardner I, Bae ON, Majid A. Comparative Cerebroprotective Potential of d- and l-Carnosine Following Ischemic Stroke in Mice. Int J Mol Sci. 2020 Apr 26;21(9):3053. doi: 10.3390/ijms21093053. PMID: 32357505; PMCID: PMC7246848.
28. Kim ES, Kim D, Nyberg S, Poma A, Cecchin D, Jain SA, Kim KA, Shin YJ, Kim EH, Kim M, Baek SH, Kim JK, Doeppner TR, Ali A, Redgrave J, Battaglia G, Majid A, Bae ON. LRP-1 functionalized polymersomes enhance the efficacy of carnosine in experimental stroke. Sci Rep. 2020 Jan 20;10(1):699. doi: 10.1038/s41598-020-57685-5. PMID: 31959846; PMCID: PMC6971073.
29. Schön M, Mousa A, Berk M, Chia WL, Ukropec J, Majid A, Ukropcová B, de Courten B. The Potential of Carnosine in Brain-Related Disorders: A Comprehensive Review of Current Evidence. Nutrients. 2019 May 28;11(6):1196. doi: 10.3390/nu11061196. PMID: 31141890; PMCID: PMC6627134.
30. Davis CK, Laud PJ, Bahor Z, Rajanikant GK, Majid A. Systematic review and stratified meta-analysis of the efficacy of carnosine in animal models of ischemic stroke. J Cereb Blood Flow Metab. 2016 Oct;36(10):1686-1694. doi: 10.1177/0271678X16658302. Epub 2016 Jul 8. PMID: 27401803; PMCID: PMC5046161.
31. Baek SH, Noh AR, Kim KA, Akram M, Shin YJ, Kim ES, Yu SW, Majid A, Bae ON. Modulation of mitochondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage. Stroke. 2014 Aug;45(8):2438-2443. doi: 10.1161/STROKEAHA.114.005183. Epub 2014 Jun 17. PMID: 24938837; PMCID: PMC4211270.
32. Bae ON, Majid A. Role of histidine/histamine in carnosine-induced neuroprotection during ischemic brain damage. Brain Res. 2013 Aug 21;1527:246-54. doi: 10.1016/j.brainres.2013.07.004. Epub 2013 Jul 11. PMID: 23850642.
33. Bae ON, Serfozo K, Baek SH, Lee KY, Dorrance A, Rumbeiha W, Fitzgerald SD, Farooq MU, Naravelta B, Bhatt A, Majid A. Safety and efficacy evaluation of carnosine, an endogenous neuroprotective agent for ischemic stroke. Stroke. 2013 Jan;44(1):205-12. doi: 10.1161/STROKEAHA.112.673954. Epub 2012 Dec 18. PMID: 23250994; PMCID: PMC3678096.
34. Min J, Senut MC, Rajanikant K, Greenberg E, Bandagi R, Zemke D, Mousa A, Kassab M, Farooq MU, Gupta R, Majid A. Differential neuroprotective effects of carnosine, anserine, and N-acetyl carnosine against permanent focal ischemia. J Neurosci Res. 2008 Oct;86(13):2984-91. doi: 10.1002/jnr.21744. PMID: 18543335; PMCID: PMC2805719.
35. Rajanikant GK, Zemke D, Senut MC, Frenkel MB, Chen AF, Gupta R, Majid A. Carnosine is neuroprotective against permanent focal cerebral ischemia in mice. Stroke. 2007 Nov;38(11):3023-31. doi: 10.1161/STROKEAHA.107.488502. Epub 2007 Oct 4. PMID: 17916766.
Methocarbamol is a CNS depressant indicated with rest, physical therapy and other treatments to control the discomfort associated with various acute musculoskeletal conditions.
Methocarbamol was developed in the early 1950s as a treatment for muscle spasticity and the associated pain.6,7 It is a guaiacol glyceryl ether.7
Methocarbamol tablets and intramuscular injections are prescription medicines indicated in the United States as an adjunct to rest, physical therapy, and other measures for the relief of discomforts associated with acute, painful musculoskeletal conditions.Label,9 In Canada, methocarbamol can be sold as an over the counter oral medicine at a lower dose that may be combined with acetaminophen or ibuprofen.10 A combination product with acetylsalicylic acid and codeine is available in Canada by prescription.10
Common side effect include headaches, sleepiness, and dizziness.[3][8] Serious side effects may include anaphylaxis, liver problems, confusion, and seizures.[4] Use is not recommended in pregnancy and breastfeeding.[3][4] Because of risk of injury, skeletal muscle relaxants should generally be avoided in geriatric patients.[3] Methocarbamol is a centrally acting muscle relaxant.[3] How it works is unclear, but it does not appear to affect muscles directly.[3]
Methocarbamol was approved for medical use in the United States in 1957.[3] It is available as a generic medication.[3][4] It is relatively inexpensive as of 2016.[9] In 2019, it was the 136th most commonly prescribed medication in the United States, with more than 4 million prescriptions.[10][11]
SYN
CN 109970606
SYN
Synthesis of methocarbamol from guaifenesin. (a) methocarbamol and (b) β-isomer of methocarbamol.
The muscle relaxant methocarbamol 2 and tranquilizer mephenoxalone 3, as well as intermediate cyclic carbonate 4, have been prepared in enantiopure form by starting from enantiopure guaifenesin 1 easily available by an entrainment resolution procedure. Thermal investigations reveal that 2 is probably a conglomerate forming substance, 3 forms a stable racemic compound, and 4 occupies an intermediate position. The enantiomeric excess of a binary phase eutectic point for these substances comprises 0%, 85%, and 10%, respectively.
Methocarbamol is a muscle relaxant used to treat acute, painful musculoskeletal spasms in a variety of musculoskeletal conditions.[12] However, there is limited and inconsistent published research on the medication’s efficacy and safety in treating musculoskeletal conditions, primarily neck and back pain.[12]
Methocarbamol injection may have a beneficial effect in the control of the neuromuscular spasms of tetanus.[6] It does not, however, replace the current treatment regimen.[6]
Currently, there is some suggestion that muscle relaxants may improve the symptoms of rheumatoid arthritis; however, there is insufficient data to prove its effectiveness as well as answer concerns regarding optimal dosing, choice of muscle relaxant, adverse effects, and functional status.[7]
Comparison to similar agents
The clinical effectiveness of methocarbamol compared to other muscle relaxants is not well-known.[12] One trial of methocarbamol versus cyclobenzaprine, a well-studied muscle relaxant, in those with localized muscle spasm found there was no significant differences in their effects on improved muscle spasm, limitation of motion, or limitation of daily activities.[12]
Contraindications
There are few contraindications to methocarbamol. They include:
Hypersensitivity to methocarbamol or to any of the injection components.[6]
While the product label states that methocarbamol can cause jaundice, there is minimal evidence to suggest that methocarbamol causes liver damage.[8] During clinical trials of methocarbamol, there were no laboratory measurements of liver damage indicators, such as serum aminotransferase (AST/ALT) levels, to confirm hepatotoxicity.[8] Although unlikely, it is impossible to rule out that methocarbamol may cause mild liver injury with use.[8]
Elderly
Skeletal muscle relaxants are associated with an increased risk of injury among older adults.[16] Methocarbamol appeared to be less sedating than other muscle relaxants, most notably cyclobenzaprine, but had similar increased risk of injury.[15][16] Methocarbamol is cited along with “most muscle relaxants” in the 2012 Beers Criteria as being “poorly tolerated by older adults, because of anticholinergic adverse effects, sedation, increased risk of fractures,” noting that “effectiveness dosages tolerated by older adults is questionable.”[17]
Pregnancy
Methocarbamol is labeled by the FDA as a pregnancy category C medication.[6] The teratogenic effects of the medication are not known and should be given to pregnant women only when clearly indicated.[6]
Overdose
There is limited information available on the acute toxicity of methocarbamol.[5][6] Overdose is used frequently in conjunction with CNS depressants such as alcohol or benzodiazepines and will have symptoms of nausea, drowsiness, blurred vision, hypotension, seizures, and coma.[6] There are reported deaths with an overdose of methocarbamol alone or in the presence of other CNS depressants.[5][6]
Abuse
Unlike other carbamates such as meprobamate and its prodrug carisoprodol, methocarbamol has greatly reduced abuse potential.[18] Studies comparing it to the benzodiazepine lorazepam and the antihistamine diphenhydramine, along with placebo, find that methocarbamol produces increased “liking” responses and some sedative-like effects; however, at higher doses dysphoria is reported.[18] It is considered to have an abuse profile similar to, but weaker than, lorazepam.[18]
The mechanism of action of methocarbamol has not currently been established.[3] Its effect is thought to be localized to the central nervous system rather than a direct effect on skeletal muscles.[3] It has no effect on the motor end plate or the peripheral nerve fiber.[6] The efficacy of the medication is likely related to its sedative effect.[3] Alternatively, methocarbamol may act via inhibition of acetylcholinesterase, similarly to carbamate.[19]
Pharmacokinetics
In healthy individuals, the plasma clearance of methocarbamol ranges between 0.20 and 0.80 L/h/kg.[6] The mean plasma elimination half-life ranges between 1 and 2 hours, and the plasma protein binding ranges between 46% and 50%.[6] The elimination half-life was longer in the elderly, those with kidney problems, and those with liver problems.[6]
Metabolism
Methocarbamol is the carbamate derivative of guaifenesin, but does not produce guaifenesin as a metabolite, because the carbamate bond is not hydrolyzed metabolically;[8][6] its metabolism is by Phase I ring hydroxylation and O-demethylation, followed by Phase II conjugation.[6] All the major metabolites are unhydrolyzed carbamates.[20][21] Small amounts of unchanged methocarbamol are also excreted in the urine.[5][6]
Society and culture
Methocarbamol was approved as a muscle relaxant for acute, painful musculoskeletal conditions in the United States in 1957.[8] Muscle relaxants are widely used to treat low back pain, one of the most frequent health problems in industrialized countries. Currently, there are more than 3 million prescriptions filled yearly.[8] Methocarbamol and orphenadrine are each used in more than 250,000 U.S. emergency department visits for lower back pain each year.[22] In the United States, low back pain is the fifth most common reason for all physician visits and the second most common symptomatic reason.[23] In 80% of primary care visits for low back pain, at least one medication was prescribed at the initial office visit and more than one third were prescribed two or more medications.[24] The most commonly prescribed drugs for low back pain included skeletal muscle relaxants.[25]Cyclobenzaprine and methocarbamol are on the U.S. Medicare formulary, which may account for the higher use of these products.[16]
Economics
The generic formulation of the medication is relatively inexpensive, costing less than the alternative metaxalone in 2016.[26][9]
Marketing
Generic methocarbamol 750mg tablet.
Methocarbamol without other ingredients is sold under the brand name Robaxin in the U.K., U.S., Canada[27] and South Africa; it is marketed as Lumirelax in France, Ortoton in Germany and many other names worldwide.[28] In combination with other active ingredients it is sold under other names: with acetaminophen (paracetamol), under trade names Robaxacet and Tylenol Body Pain Night; with ibuprofen as Robax Platinum; with acetylsalicylic acid as Robaxisal in the U.S. and Canada.[29][30] However, in Spain the tradename Robaxisal is used for the paracetamol combination instead of Robaxacet.[citation needed] These combinations are also available from independent manufacturers under generic names.[citation needed]
Research
Although opioids are a typically first line in treatment of severe pain, several trials suggest that methocarbamol may improve recovery and decrease hospital length of stay in those with muscles spasms associated with rib fractures.[31][32][33] However, methocarbamol was less useful in the treatment of acute traumatic pain in general.[34]
Long-term studies evaluating the risk of development of cancer in using methocarbamol have not been performed.[5][6] There are currently no studies evaluating the effect of methocarbamol on mutagenesis or fertility.[5][6]
The safety and efficacy of methocarbamol has not been established in pediatric individuals below the age of 16 except in tetanus.[5][6]
^ Jump up to:abcdefgh“Methocarbamol”. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. National Institute of Diabetes and Digestive and Kidney Diseases. 30 January 2017. PMID31643609.
^ Jump up to:ab See, Sharon; Ginzburg, Regina (1 August 2008). “Choosing a skeletal muscle relaxant”. American Family Physician. 78 (3): 365–70. ISSN0002-838X. PMID18711953.
^ Jump up to:abc Spence, Michele M.; Shin, Patrick J.; Lee, Eric A.; Gibbs, Nancy E. (July 2013). “Risk of injury associated with skeletal muscle relaxant use in older adults”. The Annals of Pharmacotherapy. 47 (7–8): 993–8. doi:10.1345/aph.1R735. ISSN1542-6270. PMID23821610. S2CID9037478.
^ Jump up to:abc Preston KL, Wolf B, Guarino JJ, Griffiths RR (1992). “Subjective and behavioral effects of diphenhydramine, lorazepam and methocarbamol: evaluation of abuse liability”. Journal of Pharmacology and Experimental Therapeutics. 262 (2): 707–20. PMID1501118.
^ PubChem. “Methocarbamol”. pubchem.ncbi.nlm.nih.gov. Retrieved 6 July 2020.
^ Methocarbamol. In: DRUGDEX System [intranet database]. Greenwood Village, Colorado: Thomson Healthcare; c1974–2009 [cited 2009 Feb 10].
^ Bruce RB, Turnbull LB, Newman JH (January 1971). “Metabolism of methocarbamol in the rat, dog, and human”. J Pharm Sci. 60 (1): 104–6. doi:10.1002/jps.2600600120. PMID5548215.
^ Chou, Roger; Huffman, Laurie Hoyt (2 October 2007). “Medications for Acute and Chronic Low Back Pain: A Review of the Evidence for an American Pain Society/American College of Physicians Clinical Practice Guideline”. Annals of Internal Medicine. 147 (7): 505–14. doi:10.7326/0003-4819-147-7-200710020-00008. ISSN0003-4819. PMID17909211. S2CID32719708.
^ Luo, Xuemei; Pietrobon, Ricardo; Curtis, Lesley H.; Hey, Lloyd A. (1 December 2004). “Prescription of nonsteroidal anti-inflammatory drugs and muscle relaxants for back pain in the United States”. Spine. 29 (23): E531–7. doi:10.1097/01.brs.0000146453.76528.7c. ISSN1528-1159. PMID15564901. S2CID72742439.
^ Patanwala, Asad E.; Aljuhani, Ohoud; Kopp, Brian J.; Erstad, Brian L. (October 2017). “Methocarbamol use is associated with decreased hospital length of stay in trauma patients with closed rib fractures”. The American Journal of Surgery. 214 (4): 738–42. doi:10.1016/j.amjsurg.2017.01.003. ISSN0002-9610. PMID28088301.
Sica DA, Comstock TJ, Davis J, Manning L, Powell R, Melikian A, Wright G: Pharmacokinetics and protein binding of methocarbamol in renal insufficiency and normals. Eur J Clin Pharmacol. 1990;39(2):193-4. [Article]
Bruce RB, Turnbull LB, Newman JH: Metabolism of methocarbamol in the rat, dog, and human. J Pharm Sci. 1971 Jan;60(1):104-6. [Article]
Witenko C, Moorman-Li R, Motycka C, Duane K, Hincapie-Castillo J, Leonard P, Valaer C: Considerations for the appropriate use of skeletal muscle relaxants for the management of acute low back pain. P T. 2014 Jun;39(6):427-35. [Article]
Crankshaw DP, Raper C: Mephenesin, methocarbamol, chlordiazepoxide and diazepam: actions on spinal reflexes and ventral root potentials. Br J Pharmacol. 1970 Jan;38(1):148-56. doi: 10.1111/j.1476-5381.1970.tb10343.x. [Article]
Muir WW 3rd, Sams RA, Ashcraft S: The pharmacology and pharmacokinetics of high-dose methocarbamol in horses. Equine Vet J Suppl. 1992 Feb;(11):41-4. [Article]
Authors unspecified: Methocarbamol-A New Lissive Agent. Can Med Assoc J. 1958 Dec 15;79(12):1008-9. [Article]
O’DOHERTY DS, SHIELDS CD: Methocarbamol; new agent in treatment of neurological and neuromuscular diseases. J Am Med Assoc. 1958 May 10;167(2):160-3. [Article]
A crosslinked polymeric amine that binds PHOSPHATES and BILE ACIDS; it is nonabsorbed; used for hyperphosphatemia during HEMODIALYSIS and in END-STAGE RENAL DISEASE; used like calcium acetate.
A polymeric amine that binds phosphate and is used to treat HYPERPHOSPHATEMIA in patients with kidney disease.
Drug Name:Sevelamer Carbonate,
Trade Name:Renvela®
MOA:Phosphate binder
Indication:Hyperphosphatemia
Company:Genzyme (Originator)
Sales:$1,037.9 Million (Y2015); $902.9 Million (Y2014); $997.5 Million (Y2013); $842.4 Million (Y2012); $581 Million (Y2011);ATC Code:
Sevelamer Carbonate was first approved by the U.S. Food and Drug Administration (FDA) on Jul 19, 2007, then approved by European Medicine Agency (EMA) on Jun 21, 2009. However, till 2013, China Food and Drug Administration approved this drug. It was developed by Genzyme, and the trade name is Renvela®. On the other hand, US FDA firstly approved Sevelamer HCl (Renagel®) on Oct 30, 1998.
Renvela® is a non-absorbed phosphate binding crosslinked polymer, containing multiple amines separated by one carbon from the polymer backbone. These amines exist in a protonated form in the intestine and interact with phosphate molecules through ionic and hydrogen bonding. By binding phosphate in the gastrointestinal tract and decreasing absorption, sevelamer carbonate lowers the phosphate concentration in the serum (serum phosphorus).
Renvela® is available as film-coated tablet for oral use, containing 800 mg of free sevelamer carbonate on an anhydrous basis. The initial dose is 0.8 or 1.6 grams orally three times per day with meals.SevelamerCAS Registry Number: 52757-95-6 CAS Name: 2-Propen-1-amine polymer with (chloromethyl)oxirane Additional Names: allylamine polymer with 1-chloro-2,3-epoxypropane; allylamine-epichlorohydrin copolymer; poly(allylamin-co-N,N¢-diallyl-1,3-diamino-2-hydroxypropane) Literature References: Polymeric non-absorbed phosphate binder consisting of polyallylamine crosslinked with epichlorohydrin to form a hydrogel where 40% of the amines are protonated. Follows the general formula of (C3H7N.C3H5ClO)n. Binds dietary phosphate leading to increased fecal excretion, decreased absorption and decreased serum phosphorous levels. Prepd not claimed: S. R. Holmes-Farley et al.,WO9505184; eidem,US5496545 (1995, 1996 both to GelTex). Mechanism of action: S. R. Holmes-Farley et al.,J. Macromol. Sci. Pure Appl. Chem.A36, 1085 (1999). Determn of phosphate binding capacity: J. R. Mazzeo et al.,J. Pharm. Biomed. Anal.19, 911 (1999). Clinical studies in end stage renal disease: E. A. Slatopolsky et al.,Kidney Int.55, 299 (1999). Derivative Type: Hydrochloride CAS Registry Number: 152751-57-0 Manufacturers’ Codes: PB-94; GT16-026A Trademarks: Renagel (Genzyme) Properties: Insol in water. Hydrophilic. Therap-Cat: Antihyperphosphatemic. Keywords: Antihyperphosphatemic.
Sevelamer consists of polyallylamine that is crosslinked with epichlorohydrin.[1] The marketed form sevelamer hydrochloride is a partial hydrochloride salt being present as approximately 40% aminehydrochloride and 60% sevelamer base. The amine groups of sevelamer become partially protonated in the intestine and interact with phosphate ions through ionic and hydrogen bonding.
Medical uses
Sevelamer is used in the management of hyperphosphatemia in adult patients with stage 4 and 5 chronic kidney disease (CKD) on hemodialysis. Its efficacy at lowering phosphate levels is similar to that of calcium acetate, but without the accompanying risk of hypercalcemia and arterial calcification.[2][3] In patients with CKD, it has also been shown to reduce triglycerides and LDL, and increase HDL.[4]
This is a phosphate binder.
Contraindications
Sevelamer therapy is contraindicated in hypophosphatemia or bowel obstruction. In hypophosphatemia, sevelamer could exacerbate the condition by further lowering phosphate levels in the blood, which could be fatal.[5]
Sevelamer is able to sequester advanced glycation end products (AGEs) in the gut, preventing their absorption into the blood. AGEs contribute to oxidative stress, which can damage cells (like beta cells, which produce insulin in the pancreas). As Vlassara and Uribarri explain in a 2014 review on AGEs, this may explain why sevelamer, but not calcium carbonate (a phosphate binder that does not sequester AGEs), has been shown to lower AGEs in the blood, as well as oxidative stress and inflammatory markers.[7]
Sevelamer hydrochloride (Renagel VR ) was the first polymeric phosphate control and removal of excess phosphate is of benefit to patients with chronic kidney disease CKD where dialysis is unable to maintain safe phosphorus levels. Sevelamer limits the absorption of dietary phosphorus by binding phosphate in the intestine through ionic interaction with the polyamine polymer. Sevelamer is across linked form of poly(allylamine) containing primary and secondary aliphatic amine residues and was approved for the treatment of hyperphosphatemia by the FDA in 1998.The following Scheme 5 shows the synthetic lines for Sevelamer hydrochloride.
Scheme 5. Represent the formation of poly allylamine as a residue of sevelamer structure
Synthesis of Sevelamer Hydrochloride. Approximately 40 percent of the amine moieties are in the HCl form. Crosslinking degree is 10%.
SYN
The synthesis of Sevelamer consists of crosslinkung poly(allylamine hydrochloride) with epichlorohydrin. The product is washed, dried and ground to the desired particle size to give the active substance.
The present invention relates to the process for preparation of carbonate salt of amine polymers, preferably Poly(allylamine-co-N,N′-diallyl-1,3-diamino-2-hydroxypropane)carbonate Formula-I, an antihyperphosphatemic agent.
a, b=number of primary amine groups a+b=9 c=number of crosslinking groups c=1 m=large number to indicate extended polymer network
Sevelamer carbonate is non-absorbable polymer marketed as Renvela by Genzyme Corporation. It is known chemically as poly(allylamine-co-N,N′-diallyl-1,3-diamino-2-hydroxypropane) carbonate salt. It was developed as a pharmaceutical alternative to Sevelamer hydrochloride (Renagel®). Renvela contains Sevelamer carbonate, a non-absorbed phosphate binding crosslinked polymer, free of metal and calcium. It contains multiple amines separated by one carbon from the polymer backbone. These amines exist in a protonated form in the intestine and interact with phosphate molecules through ionic and hydrogen bonding. By binding phosphate in the dietary tract and decreasing absorption, Sevelamer carbonate lowers the phosphate concentration in the serum.
[0004] Sevelamer carbonate is an anion exchange resin with the same polymeric structure as Sevelamer hydrochloride in which carbonate replaces chloride as the counterion. While the counterions differ for the two salts, the polymer itself, the active moiety, is the same. The protonated amines can be indirectly measured as carbonate content in meq/gm. Renvela is used in End Stage Renal Disease (ESRD) which leads to hyperphosphatemia due to retention of phosphorous. This condition can lead to ectopic calcification. Renvela binds dietary phosphate in GI tract and thus controls the serum phosphate levels. The potency of Renvela is measured in terms of its Phosphate Binding Capacity (PBC) by Phosphate Assay (PA). Treatment of hyperphosphatemia includes reduction in dietary intake of phosphate, inhibition of intestinal phosphate absorption with phosphate binders, and removal of phosphate with dialysis. Sevelamer carbonate taken with meals has been shown to control serum phosphorus concentrations in patients with CKD who are on dialysis. Currently Sevelamer hydrochloride is used to cure hyperphosphatemia. As a consequence ESRD patients still need a high dosage of Renagel® to meet clinical end-points, leading to adverse effect such as gastrointestinal discomfort and problems with patient compliance. But systemic acidosis development or worsening of pre-existing acidosis has been reported in many patients on long term dialysis who are given Sevelamer hydrochloride (Perit Dial Int. 2002, 22, 737-738, Nephron 2002, 92, 499-500, Kidney Int. 2004, 66, S39-S45, Ren. Fail 2005, 27,143-147).
[0005] Administration of Sevelamer hydrochloride adds to metabolic acid load because the resin removes some bicarbonate or bicarbonate precursor (mainly short chain fatty acid anions) from the body and replaces it with chloride. Each molecule of chloride contributed to the body in exchange for carbonate or bicarbonate precussor is equivalent to a molecule of hydrochloric acid added to the body, so the tendency of patients on long term haemodialysis to acidosis is inevitably increased when they take Sevelamer hydrochloride. (Kidney Int., 2005; 67: 776-777)
[0006] This problem can be countered by an increase in the dialysate concentration of bicarbonate used in each dialysis session. A more fundamental solution, suitable for both dialyzed and non-dialyzed patients, would be the administration of Sevelamer free base, or any other suitable resin, not as the chloride but as body suitable counterion such as bicarbonate. Anion exchange resins have traditionally been synthesized in the chloride form, but the chloride in the current Sevelamer preparation is of no benefit to patients with renal failure. A change in the formulation of Sevelamer from its current chloride form to Sevelamer attached to bicarbonate would convert an acid load into a mild alkali load. (Cli. Sci. 1963; 24:187-200)
[0007] U.S. Pat. No. 6,858,203 relates to phosphate-binding polymers provided for removing phosphate from the gastrointestinal tract. These polymers are useful for the treatment of hyperphosphatemia.
[0008] WO 2006/050315 describes pharmaceutical compositions comprising a carbonate salt of an aliphatic amine polymer wherein the monovalent anion can prevent or ameliorate acidosis, in particular acidosis in patients with renal disease.
[0009] HPLC Ion Chromatography PA method is used for the determination of PBC of Sevelamer HCl which can be adopted for determining the carbonate content from Sevelamer carbonate (J R Mazzeo et al, J. Pharm. Biomed. Anal. 19 (1999) 911-915).
[0010] Our co-pending application number 1402/MUM/2006 dated 1 Sep. 2006 discloses process for preparation of Sevelamer HCl having phosphate binding capacity in the range of about 5.0 meq/gm to about 6.0 meq/gm and chloride content in the range of about 3.74 to about 5.60 meq/gm.
[0011] The prior art mentioned above discussed advantages of Sevelamer carbonate over Sevelamer hydrochloride thus there remains need for commercially viable and industrially useful process for the preparation of Sevelamer carbonate having consistency in phosphate binding capacity, degree of cross linking, chloride content and carbonate content.
The reaction is represented by the following reaction scheme:
[0078] 100 gm Sevelamer hydrochloride was dispersed in 500 ml purified water and sodium hydroxide solution [20 gm sodium hydroxide dissolved in 500 ml purified water] was added to the obtained suspension followed by stirring at 25-35° C. for 30 minutes. The obtained material was filtered and wet cake was stirred in 1.0 L purified water for an hour. The material was filtered and cake was washed twice. Wet cake was dried at 50-90° C. for 5-6 hrs to get Sevelamer base (70 gm). LOD: 0.4% Chloride content: Nil.
Example 2
[0079] 10 gm Sevelamer was suspended in 200 ml water and stirred. Carbon dioxide gas was purged into the obtained suspension at 25-35° C. for 8 hrs using dry ice. The obtained material was filtered and washed with 100 ml water [3×100] and the wet cake was dried on rotavapor at 90-95° C. to get Sevelamer carbonate (11.5 gm). Yield—115% w/w [Chloride content: 0.3%, Phosphate binding: 5.75 mMole/g, Carbonate content: 4.78 meq/g and Degree of crosslinking—16.4%], Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 3
[0080] 10 gm Sevelamer was added to 200 ml water and reacted with carbon dioxide gas under pressure at 25-35° C. for 7-8 hrs with stirring. The obtained material was filtered and washed with 100 ml water thrice [3×100]. The wet cake thus obtained was dried on rotavapor at 90-95° C. to get Sevelamer carbonate (11.3 gm). Yield—113% w/w Degree of crosslinking—16.4%, Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 4
[0081] Sevelamer (7 gm) was added to 150 ml water and reacted with carbon dioxide gas under pressure at 60-65° C. for 7-8 hrs with stirring. The material obtained was filtered and washed with 100 ml purified water thrice [3×100]. The wet cake thus obtained was dried on rotavapor at 90-95° C. to get Sevelamer carbonate (9.3 gm).
[0082] Yield—120% w/w Degree of crosslinking—16.4%, Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 5
[0083] Sevelamer (7 gm) was added to 150 ml water and reacted with carbon dioxide gas by purging under pressure at 60-65° C. for 7-8 hrs with stirring. The material obtained was filtered and washed with 100 ml purified water thrice [3×100]. The wet cake thus obtained was dried on rotavapor at 90-95° C. to get Sevelamer carbonate (9.0 gm).
[0084] [Degree of crosslinking—16.4%, Chloride content: 0.5%, Phosphate binding: 5.56 mMole/g and Carbonate content: 4.46 meq/g] Yield—110% w/w Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 6
[0085] Sevelamer hydrochloride (10 gm) was treated Sodium hydroxide solution (2M) for 1 hr at temperature 25 to 35° C. to get Sevelamer base. Filter the free base and was added to 150 ml water and reacted with carbon dioxide gas by purging under pressure at 60-65° C. for 7-8 hrs with stirring. The material obtained was filtered and washed with 100 ml purified water thrice [3×100]. The wet cake thus obtained was dried on rotavapor under vacuum at 90-95° C. to get Sevelamer carbonate (9.3 gm). Yield—120% w/w.
[0086] [Degree of crosslinking—16.4%, Chloride content: 0.2%, Phosphate binding: 5.45 mMole/g and Carbonate content: 4.36 meq/g].
[0087] Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 7
[0088] Sevelamer hydrochloride (10 gm) was treated sodium hydroxide solution (2M) for 1 hr at temperature 25 to 35° C. to get Sevelamer base. Filter the free base and was added to 100 ml water. Sodium bicarbonate (10 gm dissolved in 1000 ml purified water) solution was added at temperature 60-65° C. for 4 hrs with stirring. Sevelamer Carbonate thus obtained was filtered and again subjected to for treatment of sodium bicarbonate solution (10 gm in 1000 ml). Reaction mixture was heated for 4 hrs at 60-65° C. with stirring. The material obtained was filtered and washed with 100 ml purified water thrice [3×100]. The wet cake thus obtained was dried under vacuum tray dryer at 80-90° C. for 24 hrs and further dried in atmospheric tray dryer at 100° C. for 36 hrs to get Sevelamer carbonate (9.0 gm). The loss of drying of material was about 5-7% achieved as per requirement. Yield—120% w/w, [Degree of crosslinking—16.4%, Chloride content: 0.01%, Phosphate binding: 5.68 mMole/g and Carbonate content: 4.85 meq/g]
Example 8
[0089] Sodium hydroxide pellets (41 gm) is dissolved in 600 ml methanol at 25-35° C. and polyallylamine hydrochloride (100 gm) is added to it followed by stirring for 5-6 hrs at temperature 25-35° C. The obtained reaction mass is filtered through hyflobed and filtrate is concentrated to reduce to half volume and the separated inorganic salt is filtered off over hyflobed. The obtained filtrate is concentrated completely under vacuum to get sticky mass (61 gm) of polyallylamine. Yield—61% w/w
Example 9
[0090] Polyallylamine (27.5 gm) dissolved in 100 ml water is charged into 1 L SS 316 autoclave and interacted with carbon dioxide gas under pressure (5.0 Kg/cm2). Initially 2-3 Kg/cm2 gas is consumed by the reaction mass and exotherm is observed from 28 C to 35° C. Then 5 Kg/cm2 pressure is maintained for 5-6 hours. After completion of the reaction the reaction mass is slowly added to 700 methanol and stirred for 3-4 hours. The separated solid (31 gm) is filtered, washed with 50 ml methanol and dried at 40-50° C. in vacuum oven. Yield—112% w/w
Example 10
[0091] Polyallylamine carbonate (20 gm) is dissolved in 30 ml water and cooled at 5-15° C. under stirring. The aqueous sodium hydroxide solution [dissolving 4.23 gm sodium hydroxide pellets into 4.2 ml of water] is added to reaction mass dropwise at 10-15° C. with continued stirring for 30 minutes. 101 ml toluene and 0.6 ml SPAN-85 is added to it and heated at 55-60° C. Epichlorohydrin (1.06 gm) is added to the reaction mass followed by stirring and heating for 3 hrs. The reaction mass is cooled at 25-35° C. and filtered through Buchner funnel. The obtained wet cake is added to 1 L acetone followed by stirring for 1 hour to get solid which was filtered through Buchner funnel. The aqueous organic washings are repeated for 7-10 times till polymer is free from excess alkalinity and the obtained wet cake is dried at 40-50° C. on rotavapor and then at 90-95° C. till constant weight of polymer is obtained (9 gm). Yield—45% w/w, Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
Example 11
[0092] Polyallylamine carbonate (20 gm) is dissolved in 30 ml water and cooled at 5-15° C. under stirring. The aqueous sodium hydroxide solution [dissolving 4.23 gm sodium hydroxide pellets into 4.2 ml of purified water] is added to obtained reaction mass dropwise at 10-15° C. with continued stirring for 30 minutes. 150 ml water and 0.6 ml SPAN-85 is added to it and heated at 60-80° C. Epichlorohydrin (1.06 gm) is added followed by stirring and heating is continued for 3 hours. The reaction mass is cooled at 25-35° C. and filtered through Buchner funnel. The obtained wet cake is added to 1 L acetone followed by stirring for 1 hour to get solid which is filtered through Buchner funnel. This aqueous organic washings are repeated for 7-10 times till the polymer is free from excess alkalinity and the obtained material is dried at 40-50° C. on rotavapor and/or Fluidised bed dryer then at 90-95° C. till constant weight of polymer is obtained (9 gm).
Example 12
[0093] Polyallylamine carbonate (20 gm) is dissolved in 30 ml water and cooled at 5-15° C. under stirring. The aqueous sodium hydroxide solution [dissolving 4.23 gm sodium hydroxide pellets into 4.2 ml of purified water] is added to the obtained reaction mass dropwise at 10-15° C. with continued stirring for 30 minutes. 150 ml water and 0.6 ml SPAN-85 is added to it and heated at 60-80° C. Epichlorohydrin (1.06 gm) is added followed by stirring and heating is continued for 3 hours. The reaction mass is cooled at 25-35° C. and filtered through Buchner funnel. The obtained wet cake is added to 1 L isopropyl alcohol (IPA) followed by stirring for 1 hour to get solid which is filtered through Buchner funnel. The obtained material is washed with water and organic solvents for 4-5 times till the polymer is free from excess alkalinity. The obtained wet cake is dried under vacuum tray dryer at 80-90° C. for 24 hrs and further dried in atmospheric tray dryer at 100° C. for 36 hrs till constant weight of dried polymer is obtained (15 gm). The loss on drying of material is around 6% as per requirement.
Example 13
[0094] In 1 L SS 316 autoclave, 75 gm allylamine and 200 ml water is charged and carbon dioxide gas under pressure (5 Kg/cm2) is purged into autoclave for 3-4 hours followed by stirring. Nitrogen gas is purged for 15 minutes. 9.8 gm VA-086 is added to the reaction mass and stirred at 70-80° C. for 12 hours and this solution is added to 1 L methanol under stirring. The separated material is filtered and washed with 100 ml methanol, suck dried and dried in vacuum oven at 50-60° C. to get 90 gm of polyallylamine carbonate. Yield—120% w/w
Example 14
[0095] Polyallylamine carbonate (20 gm) dissolved in 30 ml water is cooled at 5-15° C. under stirring and sodium hydroxide solution [dissolving 4.23 gm sodium hydroxide pellets into 4.2 ml of purified water] is added to the obtained reaction mass dropwise at 10-15° C. followed by continued stirring for 30 minutes. 101 ml toluene and 0.6 ml SPAN-85 is added to it and heated at 55-60° C. Epichlorohydrin (1.06 gm) is added and reaction mass is stirred and heated for 3 hours. Then it is cooled to 25-35° C. and filtered through Buchner funnel. The wet cake obtained is added to 1 to 1.5 L acetone followed by stirring for 1 hour to get solid which is filtered through Buchner funnel. The washings are repeated for 7-10 times till polymer is free from excess alkalinity. Wet cake (9 gm) is dried at 40-50° C. on rotavapor and then at 90-95° C. till constant weight of polymer is obtained. Yield—45% w/w
Example 15
[0096] Sevelamer hydrochloride (10 gm) was added to 10% aqueous sodium bicarbonate solution at 25-35° C. and stirred for 7-8 hrs. The material obtained was filtered and washed with 100 ml purified water thrice and the wet cake was dried on rotavapor at 90-95° C. to get Sevelamer carbonate (7.5 gm). Yield—75% w/w
[0097] Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate.
[0099] Sevelamer hydrochloride (10 gm) was added to 10% aqueous sodium bicarbonate solution. The mixture was stirred at 60-65° C. for 4 hrs. The material obtained was filtered and the obtained wet cake was again subjected to the treatment of 10% sodium bicarbonate solution. Reaction mixture was heated for 4 hrs at 60-65° C. with stirring. The material obtained was filtered and washed with 100 ml purified water four times and the wet cake was dried on rotavapor under vacuum at 90-95° C. to get Sevelamer carbonate (7.5 gm). Yield—75% w/w, Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate, [Chloride content: 0.03%, Phosphate binding: 5.25 mMole/g and Carbonate content: 4.65 meq/g].
Example 17
[0100] Sevelamer hydrochloride (10 gm) was added into 130 ml solution of sodium bicarbonate (10 gm NaHCO3 in 130 ml water) and the mixture was stirred at 60-65° C. for 4 hrs. The material was filtered using Buckner funnel assembly. The obtained wet cake was added into 130 ml solution of sodium bicarbonate (10 gm NaHCO3 in 130 ml water) and stirred at 60-65° C. for 4 hrs. The material was filtered using Buckner funnel assembly and the wet cake was washed by stirring it in 100 ml water for 1 hr at 60-65° C. The material was filtered using Buckner funnel assembly. The wet cake was washed twice at 60-65° C. and dried on rotavapor at 90-95° C. to get Sevelamer carbonate (8.5 gm). Yield—75% w/w, Chloride content: 0.03%
Example 18
[0101] Sevelamer hydrochloride (1.1 Kg) was added into 15.5 L solution of sodium bicarbonate (1.1 Kg NaHCO3 in 14.3 L water). The obtained mixture was stirred at 60-65° C. for 4 hrs. The obtained material was filtered by centrifuge filter. The obtained wet cake was added into 15.5 L solution of sodium bicarbonate (1.1 Kg NaHCO3 in 14.3 L water) and maintained stirring at 60-65° C. for 4 hrs. The material was filtered by centrifuge filter assembly and obtained wet cake was stirred in 11 L water for 1 hr at 60-65° C. The material was filtered by centrifuge filter and the washing of wet cake was repeated at 60-65° C. for two more times. The obtained wet cake was dried in air tray dryer (ATD) at 90-100° C. for 30-36 hrs and LOD was checked after every five hours till LOD was in the range of 5 to 10%. to get Sevelamer carbonate (0.995 Kg), [Chloride content: 0.03%, Phosphate binding capacity: 5.5 mmole/gm, Carbonate content: 5.1 meq/gm]
Example 19
[0102] Sevelamer hydrochloride (10 gm) was added to sodium bicarbonate solution (10 gm in 200 ml) at 25-35° C. The reaction mixture was heated for 4 hrs at 60-65° C. with stirring. Sevelamer Carbonate thus obtained was filtered and again subjected to treatment of Sodium bicarbonate solution (10 gm in 200 ml). Reaction mixture was heated for 4 hrs at 60-65° C. with stirring. The material was filtered off and washed with 100 ml purified water four times (4×100 ml) and the wet cake was dried under vacuum tray dryer at 80-90° C. for 24 hrs and further dried in atmospheric tray dryer at 100° C. for 36 hrs till constant weight of dried polymer was obtained. The loss on drying of material was around 6% (Limit: 4-10%), achieved as per requirement. Sevelamer carbonate (7.5 gm) was obtained which can be sieved through 30 mesh for uniformity of the sample. Yield—75% w/w. Solid state 13C NMR shows prominent peak at 164 ppm which is for carbon of carbonate. [Chloride content: 0.02%, Phosphate binding: 5.56 mMole/g and Carbonate content: 4.74 meq/g].
Example 20
[0103] 10 g wet cake of Sevelamer carbonate was subjected to drying in air tray dryer at 80-100° C. at atmospheric pressure for 36 hours and LOD was measured after every five hours. LOD: 7.5% Yield: 3.1 gm
Example 21
[0104] 100 g wet cake of Sevelamer carbonate was subjected to drying in air tray dryer at 80-100° C. at atmospheric pressure for 37 hours and LOD was measured. LOD: 8.4% Yield: 30 gm
Example 22
[0105] 10 g wet cake of Sevelamer carbonate was subjected to drying in vacuum tray dryer at 50-100° C. at reduced pressure for 24 hours and LOD was measured. LOD: 8.5% Yield: 3.2 gm
Example 23
[0106] 100 g wet cake of Sevelamer carbonate was subjected to drying in vacuum tray dryer at 50-100° C. at reduced pressure for 24 hours and LOD was measured. LOD: 8.9% Yield: 31 gm
Example 24
[0107] 10 Kg wet cake of Sevelamer carbonate was subjected to drying in fluidized bed dryer at 80-100° C. for 16 hours and LOD was measured after every five hours. LOD: 7.9% Yield: 3.4 kg
Example 25
[0108] 15 Kg wet cake of Sevelamer carbonate was subjected to drying in fluidised bed dryer at 80-110° C. for 16 hours and LOD was measured. LOD: 8.8% Yield: 4.9 kg.
Example 26
[0109] 10 g wet cake of Sevelamer carbonate was subjected to drying in rotary evaporator at 50-100° C. at reduced pressure for 16 hours and LOD was measured after every five hours.
[0110] LOD: 9.1% Yield: 3.1 gm
Example 27
[0111] 100 g wet cake of polyallylamine carbonate is subjected to drying in rotary evaporator at 50-100° C. at reduced pressure for 16 hours and LOD is measured. LOD: 8.9% Yield: 33 gm.
SYN
https://patents.google.com/patent/CN102675510A/enHyperphosphatemia is a kind of patient’s disease on one’s body that often appears at renal tubal dysfunction, hypothyroidism, acute acromegaly or phosphoric acid salt drug overdose; Its treatment is normal adopts the pharmacotherapy of regimen or oral phosphorus adsorbent to carry out; But it has been generally acknowledged that the regimen effect is relatively poor, the use of phosphorus adsorbent is essential.In recent years; Discover that the compound that contains the polyallylamine structure has good phosphorus adsorptive power (as: USP 5496545,20040191212, Chinese patent 95193521.6 etc.), crosslinked polyallylamine class medicine is evident in efficacy especially; Wherein, SEVELAMER (Sevelamer) is because of its good clinical manifestation, and granted listing is used to treat hyperphosphatemia.The structural formula of SEVELAMER is following: Synthesizing of crosslinked polyallylamine class medicine SEVELAMER (Sevelamer); Be to react by polyallylamine hydrochloride and linking agent; Different because of used linking agent, the phosphorus adsorptive power of product has than big-difference, and the most option table chloropharin of the present document of reporting is a linking agent.For example: in USP 5496545; Introduced the synthetic of a series of crosslinked allyl amine polymers oral, that the phosphorus adsorptive power is arranged, listed linking agent has: Epicholorohydrin, 1,4-butanediol diglycidyl ether, 1; 2-ethylene glycol bisthioglycolate glycidyl ether, 1; 3-propylene dichloride, 1,2-ethylene dichloride, 1,3-dibromopropane, 1; 2-ethylene dibromide, succinyl dichloride, dimethyl succinate salt, TDI, acrylate chloride and pyromellitic dianhydride, preferred cross-linking agents is an Epicholorohydrin.U.S. Pat 5496545 described SEVELAMER building-up processes are: in the alkaline aqueous solution, polyallylamine hydrochloride and Epicholorohydrin carry out crosslinking reaction under room temperature, react agglutination thing after 18 hours; Pour into and make its curing in the Virahol; Filter, repeatedly washing back redispersion is in water, after the filtration; Be scattered in a large amount of Virahols, obtain product through filtration, drying.The required reaction times of this technology is longer, and multiple times of filtration and washing in the operating process need be used a large amount of organic solvents, and in the series product, the highest phosphorus adsorptive power is 3.1mmol/g.Embodiment 1: sevelamer hydrochloride syntheticIn the 500mL flask; Add 46.2g (0.374mol) PAH hydrochloride, the 108.0mL deionized water dissolving adds sodium hydroxide and regulates pH=10-11; Drip the toluene solution of 11.0g (0.042mol) 1-chloro-3-p-toluenesulfonyl-2-propyl alcohol, heat up in 70-75 ℃ of reaction 4 hours.Reaction finishes, and adds hydrochloric acid and regulates pH=1-2, filters, and obtains the sevelamer hydrochloride bullion.The sevelamer hydrochloride bullion is scattered in the 300.0mL deionized water, and hydro-oxidation sodium is regulated pH=10.0-11.0, filters; Deionized water wash; The gained white solid through pulverizing, gets product sevelamer hydrochloride 38.8g again 70 ℃ of vacuum-dryings 8 hours; The phosphorus adsorptive value is 5.5mmol/g, and chloride ion content is 16.5%.Embodiment 2: carbonic acid SEVELAMER syntheticIn the 500mL flask; Add 150.0g (0.404mol; Weight concentration is 30%) the PAH hydrochloride aqueous solution; Add sodium hydroxide and regulate pH=10-11, drip the acetonitrile solution of 13.0g (0.042mol) 1-bromo-3-p-toluenesulfonyl-2-propyl alcohol, heat up in 70-75 ℃ of reaction 4 hours.Reaction finishes, and adds hydrochloric acid and regulates pH=1-2, filters, and obtains the sevelamer hydrochloride bullion.The sevelamer hydrochloride bullion is scattered in the 300.0mL deionized water, adds yellow soda ash and regulate pH=8.5-9.5, filter; Deionized water wash, gained white solid are 70 ℃ of vacuum-dryings 8 hours, again through pulverizing; Get product carbonic acid SEVELAMER 36.4g, the phosphorus adsorptive value is 5.4mmol/g.Embodiment 3: carbonic acid SEVELAMER syntheticIn the 500mL flask; Add 46.2g (0.374mol) PAH hydrochloride, the 108.0mL deionized water dissolving adds sodium hydroxide and regulates pH=10-11; Drip the toluene solution of 7.9g (0.042mol) 1-chloro-3-methylsulfonyl-2-propyl alcohol, heat up in 70-75 ℃ of reaction 4 hours.Reaction finishes, and adds hydrochloric acid and regulates pH=1-2, filters, and obtains the sevelamer hydrochloride bullion.The sevelamer hydrochloride bullion is scattered in the 300.0mL deionized water, and hydro-oxidation sodium is regulated pH=12.0-12.5, feeds dioxide gas to saturated; Filter; Deionized water wash, gained white solid are 70 ℃ of vacuum-dryings 8 hours, again through pulverizing; Get product carbonic acid SEVELAMER 40.5g, the phosphorus adsorptive value is 5.0mmol/g.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Clobetasol CAS Registry Number: 25122-41-2 CAS Name: (11b,16b)-21-Chloro-9-fluoro-11,17-dihydroxy-16-methylpregna-1,4-diene-3,20-dione Molecular Formula: C22H28ClFO4, Molecular Weight: 410.91 Percent Composition: C 64.30%, H 6.87%, Cl 8.63%, F 4.62%, O 15.57% Literature References: Topical corticosteroid. Prepn: Elks et al.,DE1902340; eidem,US3721687 (1969, 1973 both to Glaxo). Review of pharmacology and clinical efficacy in skin disorders: E. A. Olsen, R. C. Cornell, J. Am. Acad. Dermatol.15, 246-255 (1986). Derivative Type: 17-Propionate CAS Registry Number: 25122-46-7 Manufacturers’ Codes: GR-2/925 Trademarks: Clobesol (GSK); Dermovate (GSK); Olux (Connetics); Psorex (GSK); Temovate (GSK) Molecular Formula: C25H32ClFO5 Molecular Weight: 466.97 Percent Composition: C 64.30%, H 6.91%, Cl 7.59%, F 4.07%, O 17.13% Properties: White or almost white colorless, crystalline powder, mp 195.5-197°. [a]D +103.8° (c = 1.04 in dioxane). uv max (ethanol): 237 nm (e 15000). Insol in water. Melting point: mp 195.5-197° Optical Rotation: [a]D +103.8° (c = 1.04 in dioxane) Absorption maximum: uv max (ethanol): 237 nm (e 15000) Therap-Cat: Glucocorticoid; anti-inflammatory. Keywords: Glucocorticoid. Clobetasol propionate is a corticosteroid used to treat corticosteroid-responsive dermatoses and plaque psoriasis.
Clobetasol propionate was patented in 1968 and came into medical use in 1978.[5] It is available as a generic medication.[3] In 2019, it was the 180th most commonly prescribed medication in the United States, with more than 3 million prescriptions.[6][7]
SYNTHESIS OF KEY INTERMEDIATE
SYN
DE 1902340
US 3992422
DE 2613875
EP 72200
WO 2012122452
CN 112110972
PATENT
IN 201821008147
Clobetasol propionate (C25H32ClFO5); CAS Registry No.[25112-46-7]; IUPAC name: 17-(2′- Chloroacetyl)-9-fluoro-l l-hydroxy-10,13,16-trimethyl-3-oxo-6,7,8,l 1,12,14,15,16-octahydrocyclopenta[a]phenanthren-17-yl] propionate is a potent halogen adrenal corticosteroid of the gluco-corticoid class used to treat various skin disorders including eczema and psoriasis. It is also highly effective for contact dermatitis caused by exposure to poison ivy/oak.In the US 3721687Apatentshow use of methanesulfonyl chloride and Pyridine as base to protect alcohol and at the time of LiCI reaction results with 10-15%ene impurity and less yield.In the methanesulfonyl chloride step used with pyridine as base which is a hazardous.Mesyl compound converted to Clobetasol propionate by using LiCl in Dimethylformamide reaction at IOO-IlO0C forms 10-15% with ene impurity. The synthesis of Clobetasol propionate results in small quantities of the eneimpurity. Clobetasol propionate desired compound to be with impurities which must be minimized. Ene impurity can be reduced to very low levels by reaction itself. However, if used recrystallization reduce ene impurity it is time consuming and very expensive. Further, because recrystallizations have high losses, unacceptably low yields.
Example I: Betamethasone to betamethasone 17- propionate To a 100 ml 4-neck round bottom flask (RBF) equipped with halfmoon stirrer, thermowelland addition funnel, mounted in a tub bath, was charged betamethasone (5.0g, 0.0127mole), Dimethylformamide (20ml). Cooled the reaction mass to 10-15°C. Slowly added trimethyl ortho propionate (3.42g, 0.0255mole) and p-toluenesulfonic acid (PTSA)(0.30g, 0.00174 mole) to the reaction mass at 10-15°C. Stirred the contents 10-15°C for 4 hr. The reaction was monitored for completion by TLC. Further continued stirring at the same temperature for Ihr till reaction complies by TLC. After reaction completion, added H2SO4UP to pH=1.0-2.0 in to reaction mass.Reaction mass was quenched in Purified water (25ml) at 25-30°C. Cooled reaction mass temperature to 0-5°C. Stirred for I hr and filtered and washed with Purified water (10mlX2). Suck dried under vacuum completely to get cream coloured solid. Dried in tray drier at 50-55°C.Dry weight-5.40g(94.50%); HPLC: 98.5%;mp215-218°C. IR (KBr, on’):3454.90, 3370.99 (-OH); 1719.86, 1659.10 (C=O)iC25H33FO6;
Example 2: Betamethasone 17- propionateto betamethasone 21-tosylate 100ml 4-neck RBF equipped with halfmoon stirrer, thermowell, reflux condenser mountained in water bath, was charged Stage-1 (5.0g, O.Olllmole), Dimethylformamide (20ml). Added 4-Dimethylaminopyridine as base (4.10g, 0.0335mole) and p-toluenesulfonyl chloride (4.24.Og, 0.0222mole)slowly, Stirredfor2-3 hr at 25-30°C. Stirred reaction mass at 25-30°C till reaction complies by TLC.As such reaction mass used insitue for next step. Reaction mass aliquot taken (2ml) and quenched in DM water (20ml), precipited material fdtered and washed with DM water (20ml). Suck dried well. Dried in tray drier at 50-55°C to get dry white solid. Dry weight-0.598g, (89.0%); HPLC: 98.5%; mp-170-175°C (dec). IR (KBr,cm”1):3291.91, 2980.39 (-OH); 1739.15, 1661.99 (C=O); C32H39FO8S; MS 602.71mA 603.2317 [M+H]; 1HNMR (300MHz, CDCl3Sppm): Spectrum is recorded on Varian, and Tetra Methyl Silane (TMS) as internal standard. 1H-NMR Spectrum shows Aromatic-Ηκ7.17-7.22(d,lH); Hj-6.37-6.38 (d,lH);Hr6.14 (s,lH); HG-4.334-4.393 (m,lH);HF-3.846- 4.007(d,2H);HE-2.273-2.349 (q,2H);HD-l.671-1.688 (s,lH); Hc-I-306-1.331 (d,3H); Hb1.055-1.105 (t,3H);HA-0.941 -2.634 (m,18H).13CMR (300MHz, CDCl3Sppm): 9.055 (CH2- CH3); 17.244; 20.002; 21.353; 23.168; 27.901; 30.622; 33.508; 34.881; 36.783; 43.642; 46.637; 47.330; 48.113; 48.417; 66.613; 70.902; 93.801; 102.732; 124.415; 129.324; 130.443; 132.472; 145.632; 153.583; 168.042; 174.853 (O-C=O); 186.208 (Cyclic C=O); 205.491 (CH-CO-CH2-OAr).
Example 3:Betamethasone 21-tosylateto Clobetasol propionate As such reaction mass used insitue for next step. Added. lithium chloride (LiCl)1.04 gm (0.0245mole). Stirred the reaction mass at 60-65°C for 5-6 hr.Reaction completion checked by TLC.After reaction completion, Added DM water (200ml). Stirred the reaction mass at 10-15°C for Ihr and Filtered washed with DM water (30mlx2).Dried in oven at 50-55°C to get white crystalline powder. Dry weight-4.42gm, (85.0%); HPLC:99.70%;mp-158-161°C. IR (KBr, cm_1):3299.62, 2976.53 (-OH); 1734.32, (C=0);1662.95 (C=C);C25H32C1F05; MSΑβ6.9Ίτη/ζ 467 [M+H];’HNMR (300MHz, CDCl35ppm): Spectrum is recorded on Varian, and Tetra Methyl Silane (TMS) as internal standard. 1H-NMR Spectrum shows AromaticHk-7.094-7. 128(d,IH); Hj-6.267-6.307 (d,lH); Hr6.066-6.076 (s,IH); H0-4.334-4.393 (m,lH); Hf-3.846-4.007 (d,2H); HE-2.273-2.349 (q,2H); Hd-I .671-1.688 (s,lH); Hc-1.306- 1.331 (d,3H); Hb-I .055-1.105 (t,3H); HA-0.941-2.634 (m,17H).13CMR (300MHz, CDCl35ppm): 8.692 (CH2-CH3); 16.693; 19.664; 21.353; 23.042; 27.568; 30.387; 36.456; 41.104; 46.547; 47.422; 71.760; 93.547; 124.307; 125.728; 127.903; 129.222; 130.443; 132.472; 145.632; 153.044; 168.312; 173.101 (O-C=O); 185.802 (Cyclic C=O); 204.602(CH-CO-CH2-C1)
Synthesis of clobetasol propionate (27.1.13) starts from the known betamethasone 17-propionate (27.1.26), a potent glucocorticoid steroid with antiinflammatory and immunosuppressive properties, which was mesylated with methanesulfonyl chloride in pyridine to produce 9α-fluoro-11β-hydroxy-21-methylsulfonyloxy-16β-methyl 17-propionyloxypregna-1,4-diene-3,20-dione (27.1.27). The obtained product was refluxed in acetone, DMF, and dry LiCl mixture to produce the desired clobetasol propionate (27.1.13) [34] (Scheme 27.2.).
Clobetasol propionate, its structural formula (formula (I)), is a potent halogen-containing adrenocorticoid drug, has strong anti-inflammatory, anti-pruritic and vasoconstrictive effects, and its anti-inflammatory effect is approximately hydrogenated It is 112 times that of cortisone, and it is also used to treat neurodermatitis, contact dermatitis, eczema, discoid lupus erythematosus and other symptoms. It is currently widely used in clinical practice. It has been very popular in the international market and ranks among the top hormones. At present, there are only a few domestic companies in normal production, and the total yield is about 88%.[0003]
[0004] Formula (I).[0005] The process route for the production of synthetic clobetasol propionate is complex, technically difficult, and product quality requirements are strict. This is due to the complex structure of corticosteroids. The chemical structure of this type of drug is composed of three six-membered rings and one five-membered ring fused together to form a special molecular structure composed of 21 carbon atoms, with special molecular configuration steric effects and steric barriers. Group role. The functional groups on the drug structure interfere with each other, which makes the chemical reaction very complicated. It is manifested in many synthetic process steps, low raw material utilization rate, large amount of auxiliary materials, long production cycle, and many side reactions. The reaction process has various problems such as a large amount of solvents, a large amount of waste water and waste gas, and difficulty in recycling. Low technical indicators, low cost and other aspects.[0006] US patent, patent number 3721687, discloses two synthetic processes.[0007] Process method (1) adopts 9a-fluoro-113-hydroxy-16a-methyl-17 oxopropyl-1,4-diene-3,20-dione to synthesize clobetasol propionate, 9a -Fluoro-11-hydroxy-16 a -methyl-17oxopropyl-1,4-diene-3,20-dione and lithium chloride mixture, mixed with dimethylformamide (DMF) in acetone The solution is refluxed for four days, the solution is moved to a vacuum, ethanol, methanol, and acetone are added, and the mixture is refluxed for another 4 days. Most of the solution is moved to a vacuum, water is added to the residue, the crude product is put into the ether solution, and the mixture is passed through with chloroform. The aluminum is purified by filtration and recrystallized with ethanol to produce clobetasol propionate as a raw material. Method I uses too much acetone, and there is a certain risk of operation.[0008] Process method 2 adopts 21-chloro-9a-fluoro-1I@ -hydroxy-16a-methyl-17_oxopropyl-4ene-3,20-dione to synthesize clobetasol propionate Cable. Dissolve 21-chloro-9 a -fluoro-11 P -hydroxy-16 a -methyl-17oxopropyl_4ene-3,20-dione in acetone, cool in an ice bath, and add slowly while stirring Chromic acid (prepared by chromic acid: add 53.3ml of concentrated sulfuric acid to 250ml of water and add 66.7g of chromium trioxide); 4 hours later, the mixture reaches room temperature, ether is added, and it is left for another 20 minutes. The mixture is washed with water, and then the solution is moved to a vacuum ; The residue is recrystallized with acetone-petroleum ether, which pollutes the environment. In the past, organic solvents were not safe for production operations. [0009] Chinese patent, application number 200610053511.5, provides a method of mixing betamethasone 17-propionate sulfonate and anhydrous lithium chloride in a ratio of 1:1 to 2 and dissolving in dimethylformamide ( DMF), the chlorination reaction is carried out; second, after the chlorination reaction is complete, it is separated by ice water, and then centrifuged to dry, after drying, the crude clobetasol propionate is obtained; third, the clobetasol propionate is crude The crude tasol is dissolved in methanol or ethanol, activated carbon is added, decolorized, filtered, and the activated carbon is recovered; fourth, the filtrate is concentrated under reduced pressure, crystallized, dehydrated, and dried to obtain the raw material of clobetasol propionate. It has the characteristics of easy availability of starting materials, simple reaction steps, less dangerous and harmful solvents, mature technology, and convenient industrial production.[0010] The process route is as follows:[0011]
[0012] Clobetasol propionate uses betamethasone as the starting material, goes through the steps of cyclic ester-hydrolysis-sulfonation-chlorination, and then undergoes rough refinement to obtain clobetasol propionate-a refined substance, and then undergoes dissolution , Filtration, concentration, cooling, centrifugation, and drying to obtain clobetasol propionate. But its process route is longer, there are many influencing factors, and there are many side reactions. Moreover, the solvents used are very polluting and difficult to recycle.
Example 1[0034] 20g of Betamethasone 17-ester obtained by cyclic ester hydrolysis reaction was dissolved in 150ml of acetone, and after fully stirring and dissolving, 6g of ZnCl2 was added, and the temperature was raised to 35°C, and then 30g of BTC was introduced, After the BTC is passed, the reaction is kept warm for 3 hours. After the reaction is completed, the temperature is 40°C, and the concentration is reduced under reduced pressure until the solution contains 30ml of acetone. Then 300ml of drinking water is added for water separation and filtration. After drying for 16 hours at °C, 19.64 g of crude clobetasol propionate was obtained. The yield was 98.2%, and the crude clobetasol propionate content was 96.9% after analysis.Example 2[0036] 20g of betamethasone 17-ester compound obtained by cyclic ester hydrolysis reaction was dissolved in 150ml of acetone, and after fully stirring and dissolving, 7.2g of FeCl3 was added, heated to 30°C, and then 24g of BTC was introduced After the BTC is passed, the reaction is kept warm for 2 hours. After the reaction is completed, the solution is concentrated under reduced pressure at a temperature of 35°C until the solution contains 20ml of acetone, and then 300ml of drinking water is added for water precipitation, filtered, and finally at the temperature After drying for 10 hours at 85°C, 19.5 g of crude clobetasol propionate was obtained. The yield was 97.5%, and the crude clobetasol propionate content was 95.6% after analysis.Example 3[0038] 20g of Betamethasone 17-ester obtained by the cyclic ester hydrolysis reaction was dissolved in 150ml of acetone, and after fully stirring and dissolving, 4g of AlCl3 was added and the temperature was raised to 35°C, and then 28g of BTC was introduced, After the BTC is passed through, the reaction is kept warm for 4 hours. After the reaction is completed, the temperature is 30°C, and concentrated under reduced pressure until the solution contains 20ml of acetone. Then 300ml of drinking water is added for water precipitation, filtered, and finally at a temperature of 75 After drying for 18 hours at °C, 19.62g crude clobetasol propionate was obtained. The yield was 98.1%, and the crude clobetasol propionate content was 95.8% after analysis.Example 4[0040] The betamethasone 17-ester 20g obtained by the cyclic ester hydrolysis reaction was dissolved in 100ml of acetone, and after being fully stirred to dissolve, 4g of ZnCl3 was added, heated to 40°C, and then passed into 25g of BTC, After the BTC is passed, the reaction is kept for 5 hours. After the reaction is completed, the temperature is 40°C, and the concentration is reduced under reduced pressure until the solution contains 20ml of acetone. Then 200ml of drinking water is added for water precipitation, filtered, and finally at a temperature of 80°C. After drying for 18 hours at °C, 19.54 g of crude clobetasol propionate was obtained. The yield was 97.7%, and the crude clobetasol propionate content was 96.2% after analysis.[0041] Example 5 [0042] 20g of Betamethasone 17-ester obtained by cyclic ester hydrolysis reaction was dissolved in 200ml of acetone, and after fully stirring and dissolving, 8g of ZnCl3 was added and the temperature was raised to 50°C. Then pass in 40g BTC. After passing the BTC, keep it warm and react for 3 hours. After the reaction is completed, perform vacuum concentration at a temperature of 40°C until the solution contains 40ml of acetone, and then add 400ml of drinking water for hydrolysis. Filter, and finally dry at 85°C for 18 hours to obtain 19.58 g of crude clobetasol propionate. The yield was 97.9%, and the crude clobetasol propionate content was 96.9% after analysis.Example 6[0044] 20g of betamethasone 17-ester compound obtained by cyclic ester hydrolysis reaction was dissolved in 80ml of acetone, and after fully stirring and dissolving, 6g of ZnCl3 was added, and after the temperature was raised to 40°C, 25g of BTC was introduced, After the BTC is passed, the reaction is kept for 3 hours. After the reaction is completed, it is concentrated under reduced pressure at a temperature of 40°C, and concentrated until the solution contains 10ml of acetone. Then 150ml of drinking water is added for water precipitation, filtered, and finally at a temperature of 85°C. After drying for 18 hours at °C, 19.3g crude clobetasol propionate was obtained. The yield was 96.5%, and the crude clobetasol propionate content was 96.5% after analysis. Publication numberPriority datePublication dateAssigneeTitleUS3721687A *1968-01-191973-03-20Glaxo Lab Ltd3-keto-delta 4-9alpha-halo-11-oxygenated-16-methyl or methylene-17alpha-acyloxy-20-keto-21-halo pregnenesCN1923842A *2006-09-112007-03-07Zhejiang Dingtai Pharmaceutical Co., Ltd.Manufacturing method of clobetasol propionate Publication numberPriority datePublication dateAssigneeTitleFamily To Family CitationsCN105646630A *2015-08-102016-06-08Shandong Taihua Biological Technology Co., Ltd.One-pot Preparation of Clobetasol Propionate IntermediateCN112110972A *2019-06-212020-12-22Henan Lihua Pharmaceutical Co., Ltd.A kind of preparation method of clobetasol propionateCN112028957A *2020-07-292020-12-04Henan Lihua Pharmaceutical Co., Ltd.A kind of clobetasol propionate intermediate and preparation method
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Clobetasol propionate is used cosmetically by dark-skinned women for skin whitening, although this use is controversial. The U.S. Food and Drug Administration has not approved it for that purpose, and sales without a prescription are illegal in the U.S. Nonetheless, skin-whitening creams containing this ingredient can sometimes be found in ethnic beauty supply stores in New York City and on the internet. It is also sold internationally, and does not require a prescription in some countries. Whitening creams with clobetasol propionate, such as Hyprogel, can make skin thin and easily bruised, with visible capillaries, and acne. It can also lead to hypertension, elevated blood sugar, suppression of the body’s natural steroids, and stretch marks, which may be permanent.[10]
Clobetasol propionate is, along with mercury and hydroquinone, “amongst the most toxic and most used agents in lightening products.” Many products sold illegally have higher concentrations of clobetasol propionate than is permitted for prescription drugs.[11]
Contraindications
According to the California Environmental Protection Agency, clobetasol propionate should not be used by pregnant women, or women expecting to become pregnant soon, as studies with rats shows a risk of birth defects:[12]
“Studies in the rat following oral administration at dosage levels up to 50 mcg/kg per day revealed that the females exhibited an increase in the number of resorbed embryos and a decrease in the number of living fetuses at the highest dose. Pregnancy: Teratogenic Effects (i.e., possibility of causing abnormalities in fetuses): Pregnancy Category C: Clobetasol propionate has not been tested for teratogenicity when applied topically; however, it is absorbed percutaneously, and when administered subcutaneously it was a significant teratogen in both the rabbit and mouse. Clobetasol propionate has greater teratogenic potential than steroids that are less potent. There are no adequate and well-controlled studies of the teratogenic effects of clobetasol propionate in pregnant women. Temovate Cream and Ointment should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.”
Forms
Clobetasol propionate is marketed and sold worldwide under numerous names, including Clobex, Clob-x (Colombia), Clovate, Clobet (Biolab Thailand) Clonovate (T.O. Chemicals, Thailand), Cormax (Watson, US), Haloderm (Switzerland, by ELKO Org), Pentasol (Colombia), Cosvate, Clop (Cadila Healthcare, India), Propysalic (India), Temovate (US), Dermovate[13] (GlaxoSmithKline, Canada, Estonia, Pakistan, Switzerland, Portugal, Romania, Israel), Olux, ClobaDerm, Tenovate, Dermatovate, Butavate, Movate, Novate, Salac (Argentina), and Powercort, Lotasbat and Kloderma (Indonesia), Lemonvate (Italy), Delor (Ethiopia), Psovate (Turkey).
^ Hull C, McKeough M, Sebastian K, Kriesel J, Spruance S (March 2009). “Valacyclovir and topical clobetasol gel for the episodic treatment of herpes labialis: a patient-initiated, double-blind, placebo-controlled pilot trial”. Journal of the European Academy of Dermatology and Venereology. 23 (3): 263–7. doi:10.1111/j.1468-3083.2008.03047.x. PMID19143902. S2CID205588376.
(2S,3S)-3-{[(2Z)-2-(2-Ammonio-1,3-thiazol-4-yl)-2-{[(2-carboxy-2-propanyl)oxy]imino}acetyl]amino}-2-methyl-4-oxo-1-azetidinesulfonate2-[[(Z)-[1-(2-amino-4-thiazolyl)-2-[[(2S,3S)-2-methyl-4-oxo-1-sulfo-3-azetidinyl]amino]-2-oxoethylidene]amino]oxy]-2-methyl-propanoic acid 278-839-9[EINECS] 5159 78110-38-0[RN] UA2451400 азтреонам [Russian] [INN] أزتريونام [Arabic] [INN] 氨曲南 [Chinese] [INN] AztreonamCAS Registry Number: 78110-38-0 CAS Name: [2S-[2a,3b(Z)]]-2-[[[1-(2-Amino-4-thiazolyl)-2-[(2-methyl-4-oxo-1-sulfo-3-azetidinyl)amino]-2-oxoethylidene]amino]oxy]-2-methylpropanoic acid Additional Names: azthreonam Manufacturers’ Codes: SQ-26776 Trademarks: Azactam (BMS); Primbactam (Menarini) Molecular Formula: C13H17N5O8S2, Molecular Weight: 435.43 Percent Composition: C 35.86%, H 3.94%, N 16.08%, O 29.40%, S 14.73% Literature References: The first totally synthetic monocyclic b-lactam (monobactam) antibiotic. It has a high degree of resistance to b-lactamases and shows specific activity vs aerobic gram-negative rods. Prepn: R. B. Sykes et al.,NL8100571 (1981 to Squibb), C.A.96, 181062x (1982). Fast-atom-bombardment mass spectra: A. I. Cohen et al.,J. Pharm. Sci.71, 1065 (1982). Activity vs gram-negative bacteria: R. B. Sykes et al.,Antimicrob. Agents Chemother.21, 85 (1982). Series of articles on structure-activity, in vitro and in vivo properties, pharmacokinetics: J. Antimicrob. Chemother.8, Suppl. E, 1-148 (1981). Toxicology: G. R. Keim et al.,ibid. 141. Mechanism of action study: A. D. Russell, J. R. Furr, ibid.9, 329 (1982). Comparative stability to renal dipeptidase: H. Mikami et al.,Antimicrob. Agents Chemother.22, 693 (1982). Human pharmacokinetics: E. A. Swabb et al.,ibid.21, 944 (1982). Clinical evaluation in urinary tract infection: C. Donadio et al.,Drugs Exp. Clin. Res.13, 167 (1987). Clinical efficacy in neonatal sepsis: S. Sklavunu-Tsurutsoglu et al.,Rev. Infect. Dis.13, Suppl. 7, S591 (1991). Comprehensive description: K. Florey, Anal. Profiles Drug Subs.17, 1-39 (1988). Properties: White crystalline, odorless powder, dec 227°. Very slightly sol in ethanol, slightly sol in methanol, sol in DMF, DMSO. Practically insol in toluene, chloroform, ethyl acetate. Derivative Type: Disodium salt Molecular Formula: C13H15N5Na2O8S2, Molecular Weight: 479.40 Percent Composition: C 32.57%, H 3.15%, N 14.61%, Na 9.59%, O 26.70%, S 13.38% Properties: LD50 (mg/kg): 3300 i.v. in mice; 6600 i.p. in rats (Keim). Toxicity data: LD50 (mg/kg): 3300 i.v. in mice; 6600 i.p. in rats (Keim) Therap-Cat: Antibacterial. Keywords: Antibacterial (Antibiotics); ?Lactams; Monobactams. Aztreonam is a beta-lactam antibiotic used to treat select aztreonam sensitive gram negative bacteria.
A monocyclic beta-lactam antibiotic originally isolated from Chromobacterium violaceum. It is resistant to beta-lactamases and is used in gram-negative infections, especially of the meninges, bladder, and kidneys. It may cause a superinfection with gram-positive organisms.
Aztreonam, first discovered in 1980, is an FDA approved, intravenous, monocyclic beta-lactam antibiotic. Aztreonam is active against Gram-negative bacteria and is still used today. The oral bioavailability of aztreonam in humans is less than 1%. Herein we describe the design and synthesis of potential oral prodrugs of aztreonam.
A stirring mixture of CES1 (20 mg, 2200 Units) in a 15 mM solution of sodium phosphate monobasic (enzyme grade) in acetonitrile-d3 / D2O (1 mL; ratio of 2.5 : 97.5) was heated at 37 °C for 5 min. 2-(((Z)-(1-(2-aminothiazol-4-yl)-2-(((2S,3S)-1-((2,2- dimethyl-4-(pivaloyloxy)butoxy)sulfonyl)-2-methyl-4-oxoazetidin-3-yl)amino)-2- oxoethylidene)amino)oxy)-2-methylpropanoic acid TFA salt 28c (10 mg, 14 µmol) was added, and the suspension was stirred for 70 min at 37 °C. Over the course of the reaction a fine precipitate formed. The mixture was filtered through a 25 mm, 0.45 µM glass fiber syringe filter (Pall Corporation Acrodisc). The filtrate was analyzed by 1H-NMR spectroscopy to reveal that 3,3-dimethyltetrahydrofuran was released as one of the products. The presence of 3,3-dimethyltetrahydrofuran was confirmed by 1H-NMR analysis of the same sample spiked with 2 µL of authentic materialSYN Faming Zhuanli Shenqing, 106520857,
SYN
Synthesis Reference
Neal G. Anderson, Carl F. Anderson, “Delta form of aztreonam and preparation thereof.” U.S. Patent US4826973, issued January, 1983.
https://patents.google.com/patent/US7145017B2/enAztreonam is a monobactam antibiotic disclosed in U.S. Pat. No. 4,775,670, which is incorporated by reference herein in its entirety. Aztreonam has the chemical name (Z)-2-[[[(2-amino-4-thiazolyl)[[(2S,-3S)-2-methyl-4-oxo-1-sulfo-3-azetidinyl]carbamoyl]methylene]amino]oxy]-2-methylpropionic acid. Aztreonam is also known as [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid and (2S, 3S)-3-[[2-[2-amino-4-thiazolyl]-(Z)-2[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidine-1-sulfonic acid.Aztreonam has the structure:
Aztreonam is known to exist in various polymorphic forms including the α, β, δ, and γ forms.U.S. Pat. No. 4,775,670 discloses a process for making Aztreonam, a compound of formula I:
The process includes acylating a compound of formula IV:
The acylation entails reacting a compound of formula IV with a carboxylic acid or the corresponding carboxylic acid halide or carboxylic acid anhydride (R1—OH) in the presence of a carbodiimide such as dicyclohexylcarbodiimide and a substance capable of forming an active ester in situ such as N-hydroxybenzotriazole. U.S. Pat. No. 4,775,670 discloses that when the acyl group (R1) contains reactive functional groups, such as amino or carboxyl groups, it may be necessary to first protect those functional groups, then carry out the acylation reaction, and finally deprotect the resulting product. The deprotection is carried out by reaction of the acylation product with trifluoroacetic acid in the presence of anisole under anhydrous conditions.Similarly, U.S. Pat. No. 4,946,838 discloses a process for making crystalline anhydrous Aztreonam comprising reacting the diphenylmethyl ester of Aztreonam ([3S-[3β(Z),4α]]-3-[[(2-amino-4-thiazolyl)[(1-diphenylmethoxycarbonyl-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid) with trifluoroacetic acid in the presence of anisole under anhydrous conditions to produce the α-form of Aztreonam. The α-form is recrystallized from an anhydrous organic solvent to produce the β-form of Aztreonam. The β-form is anhydrous, substantially non-hygroscopic and more stable than the α-form.U.S. Pat. No. 5,254,681 discloses a process for preparing monobactams of formula (I):
wherein R is acyl. The process comprises acylating azetidin with 2-(2-amino-4-thiazolyl)-2-(Z)-(alkoxyimino) acetic acid in the presence of 1-hydroxy-benzotriazole and dicyclohexylcarbodiimide.U.S. Pat. No. 5,194,604 discloses a process and intermediates for making beta-lactams having aminothiazole(iminooxyacetic acid)acetic acid sidechains of formula (I), such as Aztreonam. The process comprises acylating a compound of formula III:
with a compound of formula (II):
in which R7 is
wherein
is a 4, 5, 6 or 7 membered heterocyclic ring having at least one nitrogen atom in the ring or such a group fused to a phenyl or substituted phenyl ring, to form a compound of formula (I):
wherein R1–R6 are as defined in U.S. Pat. No. 5,194,604.U.S. Pat. No. 4,652,651, which is incorporated by reference herein in its entirety, discloses a process for making 1-sulpho-2-oxoazetidine derivatives of the formula (I):
in which Het is an optionally amino-substituted, 5- or 6-membered, aromatic heterocycle containing 1 or 2 nitrogen atoms and optionally also an oxygen or sulphur atom, R1 may be lower alkoxycarbonyl-lower alkyl and R2 may be lower alkyl. The process entails acylating a compound of formula (II):
in which R20 equals R2 and R3 is hydrogen or sulpho, with a thioester of the formula (III):
in which Het is as above and R10 has any of the values of R1. U.S. Pat. No. 4,652,651 discloses that where R10 is a lower alkoxycarbonyl-lower alkyl group, for example the t-butoxycarbonylmethyl group, this can be converted, if desired, into the corresponding carboxylower alkyl group by treatment with a strong acid such as trifluoroacetic acid (optionally in the presence of anisole), hydrochloric acid or p-toluenesulphonic acid at a low temperature such as −10° C. to room temperature.There remains a need in the art for a process of making Aztreonam which does not require anhydrous reaction conditions and which also enables high yield and high purity. The present invention answers this need.
SUMMARY OF THE INVENTIONThe invention is based on the discovery that Aztreonam can be produced by reacting [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-t-butoxycarbonyl-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid with aqueous acid. The process of the invention, enables yields of between 70–75% and purities above 98%, preferably above 99%. The inventive aqueous process is advantageous over the prior art anhydrous processes in that the reaction conditions are more mild, there is no need to clean the final product and there is no need to keep the system dry. Thus, the aqueous process is less expensive than the anhydrous processes.The present invention is directed to a process for preparing [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-carboxy-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid by hydrolyzing the ester group of [3S-[3α(Z),4β]]-3-[[(2-amino-4-thiazolyl)[(1-t-butoxycarbonyl-1-methylethoxy)imino]acetyl]amino]-4-methyl-2-oxo-1-azetidinesulfonic acid. The hydrolysis may be effected by reacting the ester with aqueous acid, at elevated temperatures.One reaction scheme for carrying out the process is shown below:
EXAMPLE 15.4 g Azetidin is dissolved in 20 ml acetonitrile (or dimethyl formamide) with the assistance of 5 ml of triethylamine at room temperature. The solution is cooled to 0° C. A solution of 4 g TAEM in 25 ml THF is added with magnetic stirring. If the color disappears, 8 g TAEM in 50 ml THF is added. After 10 minutes, another 4.1 g TAEM in 25 ml THF is added. The solution is stirred at 0° C. for an additional hour. The pH is adjusted to about 4–5 with a freshly prepared TFA solution (TFA-THF 1:4, V/V). Being careful not to evaporate the acetonitrile, the THF is evaporated (weight loss is about 90 g) at 30° C. under vacuum. The remaining residue is diluted with 200 ml ethylacetate and then extracted with 100 ml and then 50 ml of distilled water. The aqueous extracts are combined and washed twice with 50 ml ethylacetate after readjustment of the pH to about 4–5. The dissolved ethylacetate is removed from the aqueous phase by vacuum at 30° C. 10–15 g KCl (or NaCl) is dissolved. The solution is acidified with HCl solution (cc. HCl-distilled water 1:4, V/V) with stirring (approx. 10 ml). The solution is cooled to 0° C. with slow stirring and crystallization occurs. The resulting suspension is refrigerated overnight (at about 5° C.). The suspension is filtered on a glass filter, and the crystals are washed with chilled water. The washed crystals are dried at room temperature. The product, Aztreonam t-butyl ester, is about 12.5–13 g white solid, which is sufficiently pure for the next step.
EXAMPLE 265 g Azetidine is dissolved in a mixture of 240 ml acetonitrile and 60 ml triethylamine. When dissolution is complete, TAEM is added in four portions. The suspension is stirred for 20–30 min, then diluted with 500 ml EtOAc and 500 ml water and stirred for 5–10 min. The pH of the emulsion is set to 5 with 2.4 M HCl solution. After the phases separate, the pH of the aqueous phase is checked. If the pH is between 4.20 and 5.30, the two phases are filtered and separated, otherwise more HCl is added. The upper phase is diluted with 900 ml ethylacetate and extracted with 2×500 ml water (faster phase separation). The combined aqueous phase is diluted with 500 ml water and washed with 2×500 ml ethylacetate. The dissolved ethylacetate is removed from the aqueous phase by vacuum. The aqueous phase is acidified further to pH 2 with 2.4 M HCl solution. The solution is stirred and cooled. Crystallization starts soon. The suspension is stirred and cooled to 0° C., stirring at this temperature overnight. The suspension is filtered, washed with chilled water, dried at 38° C. in air-circulated oven for 3 h. The yield is approx. 116–120 g of Aztreonam t-butyl ester.
EXAMPLE 3Aztreonam t-butyl ester (113.6 g, 0.231 mol) is suspended in 975 ml water at 60° C. with stirring and 325 ml trifluoroacetic acid is added. The solution is stirred for 60 min., then it is cooled slowly using an ice-water bath. After the product precipitates, the suspension is refrigerated overnight. The product is filtered on a glass-filter, suspended in 240 ml chilled water and filtered again. The filtrate is re-suspended in 360 ml cold acetone and filtered. The latter step is repeated and the product is dried at room temperature to yield 61.6 g Aztreonam (water content: 15–16%).
EXAMPLE 4Aztreonam t-butyl ester (18.0 g, 0.0366 mol) is suspended in 144 ml water at 60° C. with stirring and 40 ml aqueous hydrochloric acid (1:1, V/V) is added. The solution is stirred for 60 min, then 37 ml 5.4 M NaOH solution is added. The solution is cooled slowly using an ice-water bath. After the product precipitates, the suspension is refrigerated overnight. The product is filtered on a glass-filter, suspended in 50 ml chilled water and filtered again. The filtrate is re-suspended in 70 ml cold acetone and filtered. The latter step is repeated and the product is dried at room temperature to yield 8.3 g Aztreonam (water content: 15–16%). The crude Aztreonam is crystallized.
EXAMPLE 5Aztreonam t-butyl ester (100.00 g, Assay as is: 97.2%, 0.19796 mol)) is suspended in a mixture of 450 ml water and 5 ml trifluoroacetic acid. The suspension, which slowly becomes clear, is heated to 58° C. with stirring and 100 ml trifluoroacetic acid is added. The solution is stirred for 105 min at 60–63° C. The solution is added to chilled water (450 ml) with efficient stirring and the resulting slurry is cooled further to 25° C. After two hours it is cooled to 0° C. and stirred for 18 hours. The product is filtered on a glass-filter and washed with 300 ml chilled water. The product is suspended in 650 ml chilled water, then filtered and washed with 300 ml cold acetone. The product is suspended in 400 ml cold acetone and filtered and dried in an air-ventilation oven at 30° C. for 30 min. Yield: 66.6 g (63%, according to assays) Aztreonam (Assay: 100.5%, water content: 18.0%).HPLC Impurity Profile:
Aztreonam: 99.22%
Aztreonam t-butyl ester: 0.44% HPLC Impurity Profile of Sample from Reaction Mixture:
Aztreonam: 82.20%
Aztreonam t-butyl ester: 0.43%
Aztreonam, open-chained: 7.22%
Other main degradation product (RRT=0.56): 5.24%
EXAMPLE 6Aztreonam t-butyl ester (27.11 g, Assay as is: 96.5%, 0.05328 mol) is suspended in a mixture of 122 ml water and 1.35 ml cc. HCl. The suspension is heated to 62° C. with stirring and 30 ml cc. HCl is added. The suspension, which becomes clear after approx. 15 min, (then the product starts to precipitate), is stirred for 30 min at 63–65° C. Chilled water (162 ml) is added with efficient stirring and the resulting slurry is cooled further to 25° C. After two hours it is cooled to 0° C. and stirred for 2 hours. The product is filtered on a glass-filter, washed twice with 120 ml chilled water, twice with 125 ml cold acetone and filtered. The product is dried at room temperature overnight. Yield: 19.44 g (72%, according to assays) Aztreonam (Assay: 100.1%, water content: 14.4%).HPLC Impurity Profile:
Aztreonam: 99.65%
Aztreonam t-butyl ester: 0.21% HPLC Impurity Profile of Sample from Reaction Mixture:
Aztreonam: 89.43%
Aztreonam t-butyl ester: 0.26%
Aztreonam, open-chained: 4.70%
Other main degradation product (RRT=0.56): 1.47%
SYN
Manufacturing Process
This mixture was sterilized for 15 minutes at 121°C at 15 lbs/inch2 steam pressure prior to use. The fermentation flasks were incubated at 25°C for 40 to 45 hours on a of rotary shaker. A 250 liter batch of Agrobacterium radiobacter A.T.C.C. No. 31700 is fermented in a 100 gallon steel vessel with a media and operating conditions described below. Culture of Agrobacterium radiobacter grown out on agar slants, pH 7.3 consisted of yeast extract (1 g), beef extract (1 g), NZ amine A (2 g), glucose (10 g), agar (15 g) in 1000 ml distilled water. Loopful of surface growth from agar slant was used as the source of incolumn. Medium of oatmeal (20 g), tomato paste (20 g) tapped water to 1000 ml, pH 7, was sterilized for 15 min at 121°C at 15 lbs/inch2 steam pressure prior to use. 100 ml of the medium, containing incolumn is incubated at 25°C for about 24 hours on a rotary shaker. It was added to a mixture of yeast extract (5 g), glucose (10 g) in 1 L distilled water and incubated for about 42 hours at 25°C in 100 gallon stainless steel fermentation vessel. During incubation, the broth is agitated at 155 r.p.m. and aerated at rate of 10.0 cubic feet per minute. An antifoam agent (Ucon LB625, Union Carbide) was added as needed. The fermentation beer was adjusted to pH 4 with aqueous HCl and calls separated by centrifugation. The supernatante (200 L) was extracted with 40 L of 0.05 m cetyldimethylbenzyl ammonium chloride in dichloromethane and extract concentrated in vacuo to 5.5 L. The concentrate was then extracted with solution of 177 g of sodium thiocyanate in 2 L of water, adjusting the mixture of pH 4.35 with phosphoric acid. The aqueous extract was concentrated in vacuo to 465 ml and added to 1840 ml of methanol. Solids are filtrated yielded 194 g of crude solid product. It was dissolved and chromatographed on a 5×106.5 cm column of Sephadex G-10 three times and after concentrating in vacuo gave 3.5 g of crude antibiotic M53 (azetreonam) which was chromatographed at first on QAE Sephadex A- 25 (liner gradient, prepared from 2.5 L of water and 2.5 L of 0.25 M sodium nitrate). Then the residue (fractions 26-75) gave M53 (natrium salt) after evaporation. It was triturated with methanol and the souble fraction, 0.40 g was chromatographed on a 2.5×20 cm column of Diaion HP20AG, eluting at 2 ml per minute with water and collecting 20 ml fractions. Fractions 26-75 gave 51.9 mg of antibiotic M53 (sodium salt).
Chemical Synthesis
Aztreonam, (Z)-2[[[(2-amino-4-thiazolyl)[[(2S,3S)-2-methyl-4-oxo-1-sulfo-3-azetidinyl]cabamoyl]methylen]amino]oxy]-2-methylpropionoic acid (32.1.4.9), is synthesized from tert-butyloxycarbonylthreonine, which is reacted with O-benzylhydroxylamine in the presence of dicyclohexylcarbodimide and 1-hydroxybenzotriazole, to form the benzyl hydroxamide derivative (32.1.4.1). This product undergoes a reaction with triphenylphosphine and ethyl azodicarboxylate, which results in the cyclodehydration of the product to (3S-trans)-N-benzyloxy-3-tert-butyloxycarbonylamino-4-methyl-azetidinone (32.1.4.2). Debenzylating this by hydrogen reduction using a palladium on carbon catalyst forms (3S-trans)-N-hydroxy-3-tertbutyloxycarbonyl-amino-4-methyl-azetidinone (32.1.4.3). The hydroxyl group in this compound is removed by reducing it with titanium trichloride, which forms azetidinone (32.1.4.4). Removing the tert-butyloxycarbonyl protection using trifluoroacetic acid and subsequent acylation of the resulting product with the benzyl chloroformate gives (3S-trans)-benzyloxycarbonylamino-4-methylazetidinone (32.1.4.5). Sulfonating this product with a mixture of sulfur trioxide and dimethylformamide gives the corresponding N-sulfonic acid. Turning the resulting Nsulfonic acid into a potassium salt by reacting it with potassium hydrophosphate, followed by replacing the potassium cation with a tetrabutylammonium cation by reacting it with tetrabutylammonium sulfate gives the product (32.1.4.6). Reducing this with hydrogen using a palladium on carbon catalyst gives 3-amino-4-methyl-monobactamic acid (32.1.4.7). Acylating this with (Z) 2-amino-α-[[2-(diphenylmethoxy)-1,1-dimethyl-2-oxoethoxy]imino] 4-thiazoleacetic acid in the presence of dicyclohexylcarbodiimide and 1-hydroxy-benzotriazole gives the diphenylmethyl ester of the desired aztreonam (32.1.4.8), which is hydrolyzed to aztreonam (32.1.4.9) using trifluoroacetic acid.
It is believed that the methyl group at position 4 increases the stability of the beta-lactam ring with respect to most beta-lactamases, and at the same time it does not induce formation of beta-lactamase as cephalosporins and imipenems do.
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Aztreonam is poorly absorbed when given orally, so it must be administered as an intravenous or intramuscular injection (trade name Azactam ), or inhaled (trade name Cayston) using an ultrasonic nebulizer. In the United States, the Food and Drug Administration (FDA) approved the inhalation form on 22 February 2010, for the suppression of P. aeruginosa infections in patients with cystic fibrosis.[11] It received conditional approval for administration in Canada and the European Union in September 2009,[11] and has been fully approved in Australia.[12]
Side effects
Reported side effects include injection site reactions, rash, and rarely toxic epidermal necrolysis. Gastrointestinal side effects generally include diarrhea and nausea and vomiting. There may be drug-induced eosinophilia. Because of the unfused beta-lactam ring there is somewhat lower cross-reactivity between aztreonam and many other beta-lactam antibiotics, and it may be safe to administer aztreonam to many patients with hypersensitivity (allergies) to penicillins and nearly all cephalosporins.[13] There is a much lower risk of cross-sensitivity between aztreonam and other beta-lactam antibiotics than within other beta-lactam antibiotics. However, there is a higher chance of cross-sensitivity if a person is specifically allergic to ceftazidime, a cephalosporin. Aztreonam exhibits cross-sensitivity with ceftazidime due to a similar side chain.[14]
Mechanism of action
Aztreonam is similar in action to penicillin. It inhibits synthesis of the bacterial cell wall, by blocking peptidoglycan crosslinking. It has a very high affinity for penicillin-binding protein-3 and mild affinity for penicillin-binding protein-1a. Aztreonam binds the penicillin-binding proteins of Gram-positive and anaerobic bacteria very poorly and is largely ineffective against them.[13] Aztreonam is bactericidal, but less so than some of the cephalosporins.[medical citation needed]
^British national formulary : BNF 69 (69 ed.). British Medical Association. 2015. p. 381. ISBN9780857111562.
^World Health Organization (2019). Executive summary: the selection and use of essential medicines 2019: report of the 22nd WHO Expert Committee on the selection and use of essential medicines. Geneva: World Health Organization. hdl:10665/325773. WHO/MVP/EMP/IAU/2019.05. License: CC BY-NC-SA 3.0 IGO.
^World Health Organization (2019). The selection and use of essential medicines: report of the WHO Expert Committee on Selection and Use of Essential Medicines, 2019 (including the 21st WHO Model List of Essential Medicines and the 7th WHO Model List of Essential Medicines for Children). Geneva: World Health Organization. hdl:10665/330668. ISBN9789241210300. ISSN0512-3054. WHO technical report series;1021.
^ Kobayashi Y, Uchida H, Kawakami Y (December 1992). “Synergy with aztreonam and arbekacin or tobramycin against Pseudomonas aeruginosa isolated from blood”. The Journal of Antimicrobial Chemotherapy. 30 (6): 871–2. doi:10.1093/jac/30.6.871. PMID1289363.
Publication numberPriority datePublication dateAssigneeTitleEP0070024A11981-07-131983-01-19E.R. Squibb & Sons, Inc.The crystalline anhydrous form of (3S-(3 alpha(z),4 beta))-3-(((2-amino-4-thiazolyl)(1-carboxy-1-methylethoxy)-imino)-acetyl)-amino)-4-methyl-2-oxo-1-azetidinesulfonic acid, method for its preparation, mixture and pharmaceutical composition containing itUS4529698A1981-01-191985-07-16E. R. Squibb & Sons, Inc.Process for preparing a 2-oxo-1-azetidinesulfonic acid saltUS4652651A1983-05-311987-03-24Hoffmann-La Roche Inc.Process for the manufacture of 1-sulpho-2-oxoazetidine carboxylic acid intermediates via catalytic ester cleavageUS4775670A1980-09-291988-10-04E. R. Squibb & Sons, Inc.2-oxo-1-azetidinesulfonic acid saltsEP0297580A11987-07-011989-01-04E.R. Squibb & Sons, Inc.Amorphous form of aztreonamUS4826973A1984-07-201989-05-02E. R. Squibb & Sons, Inc.Delta form of aztreonam and preparation thereofUS4923998A1977-03-141990-05-08Fujisawa Pharmaceutical Company, Ltd.Cephem and cepham compounds and processes for preparation thereofUS4946838A1981-07-131990-08-07E. R. Squibb & Sons, Inc.Crystalline anhydrous aztreonamUS5194604A1990-06-291993-03-16E. R. Squibb & Sons, Inc.Process and intermediates for beta-lactams having aminothiazole(iminooxyacetic acid)acetic acid sidechainsUS5254681A1989-08-021993-10-19Consiglio Nazionale Delle RicercheProcess for preparing monobactames and their intermediate productPL165700B11991-10-151995-01-31PanMethod of obtaining z2/2-aminothiazolyl-4/-2/-t-butoxycarbonyl-1-methylethoxyimine/ acetic acidWO2002051356A22000-12-272002-07-04Salus Pharma, Inc.Inhalable aztreonam for treatment and prevention of pulmonary bacterial infectionsWO2003018578A12001-08-272003-03-06Aurobindo Pharma Ltd.Method for producing beta form of crystalline anhydrous aztreonamUS20040062721A12000-12-272004-04-01Montgomery Alan BruceInhalable aztreonam lysinate formulation for treatment and prevention of pulmonary bacterial infectionsWO2004052333A12002-12-112004-06-24Pari GmbhPharmaceutical compositions for the pulmonary delivery of aztreonamUS20050014739A12003-05-152005-01-20Viktor GyollaiAztreonam beta polymorph with very low residual solvent contentUS20050032775A12003-07-022005-02-10Viktor GyollaiAztreonam L-lysine and methods for the preparation thereof
Family To Family CitationsCN1802371A2003-05-152006-07-12特瓦药厂有限公司Aztreonam beta-polymorph with very low residual solvent contentAU2004256124B2 *2003-07-022011-04-28Corus Pharma, Inc.Aztreonam L-lysine and methods for the preparation thereofWO2006122253A1 *2005-05-092006-11-16Sicor, Inc.Process for making aztreonamWO2007083187A2 *2006-01-162007-07-26Orchid Chemicals & Pharmaceuticals LimitedAn improved process for the preparation of monobactam antibioticCN101412715B *2008-12-162010-04-14海南百那医药发展有限公司Aztreonam compound and preparation thereofCN102127068B *2010-12-312012-08-29山西普德药业股份有限公司Method for synthesizing aztreonam compoundCN102311431B *2011-08-302014-12-10海南海药股份有限公司Method for preparing anhydrous beta-aztreonamCN105017241B *2015-06-242018-03-06山东罗欣药业集团股份有限公司A kind of aztreonam compound and its preparation
85721-05-7, C24H27ClN2O2S, 443.0ClopenthixolCAS Registry Number: 982-24-1 CAS Name: 4-[3-(2-Chloro-9H-thioxanthen-9-ylidene)propyl]-1-piperazineethanol Additional Names: 2-chloro-9-[3-[4-(2-hydroxyethyl)-1-piperazinyl]propylidene]thiaxanthene Molecular Formula: C22H25ClN2OS Molecular Weight: 400.96 Percent Composition: C 65.90%, H 6.28%, Cl 8.84%, N 6.99%, O 3.99%, S 8.00% Literature References: Thioxanthene neuroleptic. Prepn (configuration not specified): BE585338; P. V. Petersen et al.,US3116291 (1960, 1963 both to Kefalas A/S). Prepn of the pharmacologically active cis-isomer: BE816855; N. Lassen, US3996211 (1974, 1976 both to Kefalas A/S). Pharmacology: Cazzullo, Andreola, Acta Neurol.20, 162 (1965); Weissman, Mod. Probl. Pharmacopsychiatry2, 15 (1969); Moeller Nielsen, ibid. 23. Metabolism: Khan, Acta Pharmacol. Toxicol.27, 202 (1969). HPLC determn of isomers in serum: T. Aaes-Jorgensen, J. Chromatogr.188, 239 (1980). Series of articles on pharmacology and clinical studies: Acta Psychiatr. Scand.64, Suppl. 294, 1-77 (1981). Properties: Colorless syrup. Sparingly sol in ether. Readily sol in methanol. Derivative Type: Dihydrochloride CAS Registry Number: 633-59-0 Manufacturers’ Codes: AY-62021; N-746 Trademarks: Ciatyl (Troponwerke); Sordenac (Lundbeck); Sordinol (Ayerst) Molecular Formula: C22H25ClN2OS.2HCl Molecular Weight: 473.89 Percent Composition: C 55.76%, H 5.74%, Cl 22.44%, N 5.91%, O 3.38%, S 6.77% Properties: Crystals from ethanol, mp 250-260° (dec). Freely sol in water; sparingly sol in alcohol. Practically insol in other organic solvents. LD50 in male mice (mg/kg): 111 i.v. (Lassen). Melting point: mp 250-260° (dec) Toxicity data: LD50 in male mice (mg/kg): 111 i.v. (Lassen)
Derivative Type:cis(Z)-Form dihydrochloride CAS Registry Number: 58045-23-1 Trademarks: Cisordinol (Lundbeck); Clopixol (HMR) Properties: Crystals, mp 250-260° (dec). LD50 in male mice (mg/kg): 105 i.v. (Lassen). Melting point: mp 250-260° (dec) Toxicity data: LD50 in male mice (mg/kg): 105 i.v. (Lassen) Therap-Cat: Antipsychotic. Keywords: Antipsychotic; Thioxanthenes. Zuclopenthixol is an antipsychotic indicated for the management of schizophrenia. The acuphase formulation is indicated for initial treatment of acute psychosis or exacerbation of psychosis, while the depot formulation is best for maintenance.Zuclopenthixol, also known as Zuclopentixol or Zuclopenthixolum, is an antipsychotic agent. Zuclopenthixol is a thioxanthene-based neuroleptic with therapeutic actions similar to the phenothiazine antipsychotics. It is an antagonist at D1 and D2 dopamine receptors. Major brands of zuclopenthixol are Cisordinol, Acuphase, and Clopixol. This drug is a liquid. This compound belongs to the thioxanthenes. These are organic polycyclic compounds containing a thioxanthene moiety, which is an aromatic tricycle derived from xanthene by replacing the oxygen atom with a sulfur atom. Known drug targets of zuclopenthixol include 5-hydroxytryptamine receptor 2A, D(1B) dopamine receptor, D(2) dopamine receptor, D(1A) dopamine receptor, and alpha-1A adrenergic receptor. It is known that zuclopenthixol is metabolized by Cytochrome P450 2D6. Zuclopenthixol was approved for use in Canada in 2011, but is not approved for use in the United States.
Zuclopenthixol (brand names Cisordinol, Clopixol and others), also known as zuclopentixol, is a medication used to treat schizophrenia and other psychoses. It is classed, pharmacologically, as a typical antipsychotic. Chemically it is a thioxanthene. It is the cis–isomer of clopenthixol (Sordinol, Ciatyl).[1] Clopenthixol was introduced in 1961, while zuclopenthixol was introduced in 1978.
Zuclopenthixol is a D1 and D2 antagonist, α1-adrenergic and 5-HT2 antagonist.[2] While it is approved for use in Australia, Canada, Ireland, India, New Zealand, Singapore, South Africa and the UK it is not approved for use in the United States.[3][4]
Medical uses
Available forms
Zuclopenthixol is available in three major preparations:
As zuclopenthixol decanoate (Clopixol Depot, Cisordinol Depot), it is a long-acting intramuscular injection. Its main use is as a long-acting injection given every two or three weeks to people with schizophrenia who have a poor compliance with medication and suffer frequent relapses of illness.[5] There is some evidence it may be more helpful in managing aggressive behaviour.[6]
As zuclopenthixol acetate (Clopixol-Acuphase, Cisordinol-Acutard), it is a shorter-acting intramuscular injection used in the acute sedation of psychotic inpatients. The effect peaks at 48–72 hours providing 2–3 days of sedation.[7]
As zuclopenthixol dihydrochloride (Clopixol, Cisordinol), it is a tablet used in the treatment of schizophrenia in those who are compliant with oral medication.[8]
It is also used in the treatment of acute bipolar mania.
Dosing
As a long-acting injection, zuclopenthixol decanoate comes in a 200 mg and 500 mg ampoule. Doses can vary from 50 mg weekly to the maximum licensed dose of 600 mg weekly. In general, the lowest effective dose to prevent relapse is preferred. The interval may be shorter as a patient starts on the medication before extending to 3 weekly intervals subsequently. The dose should be reviewed and reduced if side effects occur, though in the short-term an anticholinergic medication benztropine may be helpful for tremor and stiffness, while diazepam may be helpful for akathisia. 100 mg of zuclopenthixol decanoate is roughly equivalent to 20 mg of flupentixol decanoate or 12.5 mg of fluphenazine decanoate.
In acutely psychotic and agitated inpatients, 50 – 200 mg of zuclopenthixol acetate may be given for a calming effect over the subsequent three days, with a maximum dose of 400 mg in total to be given. As it is a long-acting medication, care must be taken not to give an excessive dose.
In oral form zuclopenthixol is available in 10, 25 and 40 mg tablets, with a dose range of 20–60 mg daily.
Side effects
Chronic administration of zuclopenthixol (30 mg/kg/day for two years) in rats resulted in small, but significant, increases in the incidence of thyroid parafollicular carcinomas and, in females, of mammary adenocarcinomas and of pancreatic islet cell adenomas and carcinomas. An increase in the incidence of mammary adenocarcinomas is a common finding for D2 antagonists which increase prolactin secretion when administered to rats. An increase in the incidence of pancreatic islet cell tumours has been observed for some other D2 antagonists. The physiological differences between rats and humans with regard to prolactin make the clinical significance of these findings unclear.
Withdrawal syndrome: Abrupt cessation of therapy may cause acute withdrawal symptoms (eg, nausea, vomiting, or insomnia). Symptoms usually begin in 1 to 4 days of withdrawal and subside within 1 to 2 weeks.[1][2]
Other permanent side effects are similar to many other typical antipsychotics, namely extrapyramidal symptoms as a result of dopamine blockade in subcortical areas of the brain. This may result in symptoms similar to those seen in Parkinson’s disease and include a restlessness and inability to sit still known as akathisia, a slow tremor and stiffness of the limbs.[8] Zuclopenthixol is thought to be more sedating than the related flupentixol, though possibly less likely to induce extrapyramidal symptoms than other typical depots.[5] As with other dopamine antagonists, zuclopenthixol may sometimes elevate prolactin levels; this may occasionally result in amenorrhoea or galactorrhoea in severe cases. Neuroleptic malignant syndrome is a rare but potentially fatal side effect. Any unexpected deterioration in mental state with confusion and muscle stiffness should be seen by a physician.
Zuclopenthixol decanoate induces a transient dose-dependent sedation. However, if the patient is switched to maintenance treatment with zuclopenthixol decanoate from oral zuclopenthixol or from i.m. zuclopenthixol acetate the sedation will be no problem. Tolerance to the unspecific sedative effect develops rapidly.[9]
SYN
Journal of the American Chemical Society (2019), 141(6), 2251-2256
Inside a nitrogen-filled glovebox, an oven-dried glass culture tube (Fischer Scientific part #14- 959-35A), equipped with a magnetic stirring bar, was charged with 2-chloro-9H-thioxanthen-9- one (245 mg, 1.0 mmol, 1 equiv), copper(II) acetate (0.91 mg, 0.0050 mmol, 0.0050 equiv), racBINAP (3.2 mg, 0.0050 mmol, 0.0050 equiv), and THF (1.0 mL). The tube was then fitted tightly with a Teflon-lined blow-out screw cap (Kimble-Chase part #73808-15425). The reaction tube was removed from the glovebox, and the mixture was stirred rapidly for 5 min. A balloon, connected to a 6 mL plastic syringe head, was filled with allene gas until its size was roughly 6 cm in diameter. A needle was attached to the head of the syringe. The reaction tube was evacuated by piercing the septum with a needle connected to a Schlenk line. Immediately after, the allene contained in the balloon was used to refill the reaction tube by piercing the septum with the needle. The balloon decreased to roughly half its original diameter during the refill process. The needle and balloon were left attached, and dimethoxy(methyl)silane (250 uL, 2.0 mmol, 2.0 equiv) was added to the reaction mixture using a 1 mL plastic syringe. The solution was then stirred overnight at rt. At this point, the flask was quickly evacuated by piercing the septum with a needle connected to a Schlenk line, and the headspace was refilled with dry nitrogen. This process was repeated a total of three times. THF (1 mL) solution containing 4e (367 mg, 1.2 mmol, 1.2 equiv), triphenylphosphine (11.5 mg, 0.044 mmol, 0.044 equiv), racDTBM-SEGPHOS (26.8 mg, 0.044 mmol, 0.044 equiv), and copper(II) acetate (3.6 mg, 0.040 mmol, 0.040 equiv) was added to the reaction mixture using a 1 mL plastic syringe. The reaction tube was heated to 40 °C by submersion in an oil bath overnight. After cooling to rt, the cap was removed and 4 M HCl in dioxane was slowly added to the reaction mixture (2.0 mL, WARNING: VIGOROUS HYDROGEN GAS EVOLUTION). The color of the reaction mixture turned to deep red, and after stirring for approximately 30 min, a tan precipitate evolved. After an additional 1 h, diethyl ether (10 mL) was added and the solids collected by filtration (950 mg). By LC/MS analysis, this solid contains mostly 4c (as the hydrochloride) with a trace amount of triphenylphosphine oxide. The entire solid was suspended in dry acetonitrile (1.0 mL) in another dry reaction tube, equipped with a magnetic stirring bar. Potassium carbonate (552 mg, 4 mmol) was added to the tube, which was then capped and placed under a nitrogen atmosphere using a needle connected to a Schlenk line. 2-bromoethanol (142 uL, 2 mmol) was added to the reaction mixture using a glass microsyringe, and the mixture was left to stir overnight at rt. After this time, the cap was removed, and the solution was diluted with water (10 mL). The mixture was extracted with dichloromethane (3 x 10 mL), and the combined organic phases was concentrated with the aid of a rotary evaporator. The mixture was purified by reverse phase preparative HPLC (C18 column, MeCN/water) to yield a 1.1:1 Z/E mixture of 4d as a yellow foamy solid (217 mg, 54% overall yield). The identity of 4d was confirmed by LC/MS analysis against a commercially available standard (Cayman Chemical) and by comparison of 1H NMR to the literature. 13 For further structural confirmation, a portion of 4d was repurified by HPLC to obtain pure (Z)-4d, the biologically active isomer, whose spectra have not been reported in the literature. 1H NMR (400 MHz, CDCl3) δ 7.43 (dd, J = 14.5, 6.2 Hz, 3H), 7.27 (q, J = 8.2 Hz, 4H), 7.17 (d, J = 8.3 Hz, 1H), 5.89 (t, J = 7.1 Hz, 1H), 3.60 (t, J = 5.4 Hz, 2H), 2.62 (t, J = 7.2 Hz, 2H), 2.58–2.35 (m, 12H); 13C NMR (101 MHz, CDCl3) δ 140.2, 135.7, 133.4, 133.2, 132.7, 130.9, 130.4, 128.6, 127.3, 126.9, 126.8, 126.7, 126.2, 125.6, 59.2, 58.3, 57.7, 53.1, 52.8, 27.3.
SYN
Chemical Engineering & Technology (2016), 39(10), 1821-1827.
https://patents.google.com/patent/CN103214453A/enDiuril ton (Clopenthixol), chemistry 2-chloro-9-[3 ‘ by name-(N ‘-the 2-hydroxyethyl piperazine-N)-allyl group]-thioxanthene, this product is a kind of Thiaxanthene derivative, has significant antipsycholic action and special sedative effect, is particularly useful for the schizophrenia patient.Its activeconstituents is its alpha-isomer, i.e. zuclopenthixol (structural formula is seen Fig. 1); Have the stereotypy effect that anti-Ritalin causes, and the effect of anti-Apomorphine is arranged, this product energy rejection condition avoiding reaction and catalepsy are stronger 10 times than chlorpromazine.A little less than the cholinolytic effect, and antihistamine effect is strong.Zuclopenthixol is applicable to that treatment has psychosis, class Paranoia-illusion type schizophrenia, hebephrenia, the manic and anxiety periodic psychosis of anxiety and illusion symptom; The uneasiness that mental element causes, excitement, psychiatric disorder, the encephalatrophy process, post-traumatic psychosis, the proverb of trembling are absurd etc.Be particularly useful for elderly patients.Recorded the quality standard of zuclopenthixol sheet, zuclopenthixol dihydrochloride, Ciatyl Depot and zuclopenthixol acetic ester etc. in the British Pharmacopoeia, wherein the zuclopenthixol quality standard has stipulated that its content should be 95%-105%.But in actual industrial production, guarantee that zuclopenthixol reaches pharmaceutically acceptable purity, and β-isomer (structural formula is seen Fig. 2) content being limited in 5%, is a very thing of difficulty.About the preparation method of zuclopenthixol, mainly containing of bibliographical information is following several:As if the general separation of having described diuril ton isomer can be undertaken by the fractional crystallization of dihydrochloride among the BE585338A of nineteen fifty-nine application, and still, this separation method yield is extremely low, and complicated operation does not also have actual industrial use.Described the preparation method of diuril ton isomer mixture among the US3116291, wherein alpha-isomer is that the content of zuclopenthixol is 30%-35%.Obtain purer zuclopenthixol by diuril ton alkali being carried out fractional separation in the literary composition with ether organic solvent, but, instructed crystallization to come the purifying zuclopenthixol can not obtain good result, especially in isomer mixture, had under the situation of a large amount of impurity existence by diuril ton alkali.Embodiment 1: the preparation of diuril ton base1) preparation of 2-chloro-9-allyl group-9-thioxanthene alcohol100.00g (0.405mol) 2-chloro-9-thioxanthone is dissolved in the 600mL tetrahydrofuran (THF), 20 ℃ of-30 ℃ of stirrings, add magnesium powder 26g then, iodine 1g, splash into chlorallylene 65g (0.855mol), 40 ℃-50 ℃ are reacted 2h down, and the cooling back drips 20% sodium chloride aqueous solution 1000ml in reaction solution, stir 10min, filter insolubles, use dichloromethane extraction then 2 times, each 500ml, merge organic phase, water 500ml washing is told organic layer, dry after-filtration, filtrate is concentrated except that desolvating, obtain 105.40g2-chloro-9-allyl group-9-thioxanthene alcohol.2) preparation of 2-chloro-9-(2-propenylidene) thioxanthene100.00g (0.346mol) 2-chloro-9-allyl group-9-thioxanthene alcohol is dissolved in the 100ml toluene, solution is heated to 40 ℃, the Acetyl Chloride 98Min. of 1.34g (0.017mol) is dissolved in the diacetyl oxide of 41.19g (0.403mol) and drops in the above-mentioned solution, temperature is controlled at about 40 ℃, dropwise, it is complete until the TLC monitoring reaction that heating makes temperature of reaction rise to 50 ℃ of-55 ℃ of reactions, and concentrating under reduced pressure steams solvent, obtains 94.01g2-chloro-9-(2-propenylidene) thioxanthene.3) preparation of clopenthixol baseN-(2-hydroxyethyl) piperazine of getting 90.00g (0.332mol) 2-chloro-9-(2-propenylidene) thioxanthene and 215.21g (1.65mol) adds in the 1L four-hole boiling flask, stirs and is warming up to 100 ℃ of reactions, and TLC monitors to reacting completely.Vacuum oil pump concentrating under reduced pressure excessive N-(2-hydroxyethyl) piperazine, temperature is controlled at 100 ℃-135 ℃, and oil pump vacuum tightness is at 0.2-1mmHg.Distillation finishes, and adds the benzene of 400ml and the water of 100ml in gained oily matter, and 70 ℃ are stirred 15min, and separatory is used the water washing organic phase of 100ml again, and simultaneous temperature is controlled at 60 ℃-70 ℃, separatory; With organic phase concentrate resistates.This resistates is dissolved in the 300ml methylene dichloride, after add 10% hydrochloric acid soln and transfer pH to 2-3, stirring 10min, separatory, water discard dichloromethane extraction liquid with the dichloromethane extraction of 150ml; Above-mentioned water adds ammoniacal liquor and regulates pH=9-10, extract with methylene dichloride (300ml * 2) after stirring 10min, merge organic phase, use anhydrous sodium sulfate drying, suction filtration, filtrate decompression concentrate 109.32g diuril ton base, isomer proportion α/β is that 45/55 (the HPLC area normalization method: analytical column is 4.6 * 250mmAgilent C18 post, moving phase is acetonitrile: methyl alcohol: phosphoric acid buffer=20: 30: 50, flow velocity are 1mL/min; With this understanding, the retention time of zuclopenthixol is 12min, and the retention time of β-isomer is 16min).Embodiment 2: the preparation of zuclopenthixol Chlorodracylic acid ester 2HCl100.00g (0.250mol) α/β-diuril ton is dissolved in the ethyl acetate of 500ml, under 40 ℃ of conditions, drips the 100ml ethyl acetate that is dissolved with 52.48g (0.30mol) parachlorobenzoyl chloride, dropwise the back back flow reaction, complete until the TLC monitoring reaction.Remove the 300ml solvent under reduced pressure, be cooled to 4 ℃, remove by filter precipitation.Mother liquor is heated to 40 ℃, drips the concentrated hydrochloric acid aqueous solution of 12.50g (0.125mol) 37%, react about 1h after, cool off, have solid to separate out, filter 56.27g zuclopenthixol Chlorodracylic acid ester 2HCl.Purity (HPLC is the same) is 97.11%, and productive rate is 36.82%.Embodiment 3: the preparation of zuclopenthixol2HCl is dissolved in the methanol aqueous solution of 300ml80% with 45.25g (0.074mol) zuclopenthixol Chlorodracylic acid ester, adds the potassium hydroxide of 16.83mol (0.30mol) then.With mixture heating up to 50 ℃, insulation reaction 1h.Underpressure distillation removes and desolvates, and with toluene (200ml * 2) and water extraction, merges organic phase, and concentrating under reduced pressure is removed toluene; Residue obtainedly carry out recrystallization with hexanaphthene, the 25.51g dried crystals.Purity is 99.7%, and productive rate is 86.17%. 1H?NMR(CDCl 3,400MHz),δ:7.10-7.46(7H,m),5.98(1H,t),3.41(2H,t),2.46-2.52(14H,m)。Embodiment 4: the preparation of Ciatyl Depot 2HClThe zuclopenthixol of 50.00g (0.125mol) is dissolved in the methylene dichloride of 500ml, and to wherein dripping 28.60g (0.150mol) decanoyl chloride, back flow reaction is complete to the TLC monitoring reaction after dropwising under the room temperature.Underpressure distillation removes and desolvates, and adds the ethyl acetate of 300ml in residue, drips the ethyl acetate solution that contains hydrogenchloride again, is transferred to 3-4 until pH.After the cooling, filter, vacuum-drying gets 70.63g Ciatyl Depot 2HCl.Productive rate is 90.12%.Embodiment 5: the preparation of Ciatyl DepotThe Ciatyl Depot 2HCl of 60g (0.0957mol) is suspended in the t-butyl methyl ether of 400ml, drips water (250ml) solution of 13.22g (0.0957mol) salt of wormwood, stirring reaction 0.5h.Two are separated, and use 100ml water washing organic phase again, use the anhydrous sodium sulfate drying organic phase, filter, and organic solvent is removed in underpressure distillation, get the 51.29g Ciatyl Depot.Productive rate is 96.58%. 1H?NMR(CDCl 3,400MHz),δ:7.12-7.50(7H,m),5.90(1H,t),4.35(2H,t),3.41(2H,t),2.97(2H,t),2.32-2.57(10H,m),2.06(2H,t),1.64(2H,m),1.30(12H,m),0.88(3H,t)。
ZU3To a solution of 9-oxothioxanthene (1.0 equiv.) in THF at reflux were added a solution of cyclopropylmagnesium bromide in THF (1.0 equiv.) and stirred during 2 hours. The mixture was cooled down at room temperature and a solution of hydrogen bromide in acetic acid (4 eq.) was added and stirred at room temperature. The reaction mixture was concentrated in vacuo and purified by silica gel column chromatography to obtain 9- (3bromopropylidene)thioxanthene in 30% yield.Then, to a solution of 9-(3bromopropylidene)thioxanthene in acetonitrile at reflux was added N-(2-hydroxyethyl)piperazine (1,5 eq.), potassium iodide (0.1 eq.) and potassium carbonate (3 eq.). The mixture was stirred at reflux then concentrated in vacuo and purified by silica gel column chromatography to afford ZU3 with 98% purity (HPLC). HPLC analysis (BEH C18 type, mobile phase: H20/ acetonitrile (HCOOH 0.1%)) : tR = 1.68 min. Preparation of ZUf:l-(3-(9H-thioxanthen-9-ylidene)propyl)piperidine-4-carboxylic acid), ZU4 (9-(3-(4- (ethylacetate) iperidine)propylidene)-thioxanthene
To a solution of 9-(3-bromopropylidene)thioxanthene in acetonitrile at reflux was added N-ethylacetate piperidine (1,5 eq.), potassium iodide (0.1 eq.) and potassium carbonate (3 eq.). The mixture was stirred at reflux then concentrated in vacuo and purified by silica gel column chromatography to afford ZU4 as a brown oil with a purity of 97% in HPLC analysis HPLC analysis (BEH C18 type, mobile phase: H20/acetonitrile (HCOOH 0.1%)) : tR = 2.07 min.The compound ZU4 was stirred during 2 hours at reflux in a mixture of THF and a solution of NaOH in water. After phase separation, the aqueous layer was extracted twice by diethyl ether. The global organic layer was, then, washed by a saturated solution of NaCl, dried over MgSC^, filtered and concentrated in vacuo to afford ZUf HPLC analysis (BEH CI 8 type, mobile phase: H20/acetonitrile (HCOOH 0.1%)) : tR = 2.24 min.Preparation of ZU5:(Z)-2-(4-(3-(2-chloro-9H-thioxanthen-9-ylidene)propyl)piperazin-l-yl)ethylacetate
To a solution of ZU (4-[3-(2-chloro-9H-thioxanthen-9-ylidene)propyl]-l- piperazineethanol) (leq.) in dichloromethane was added acetic anhydride (1.5 eq.), 4- dimethylaminopyridine (0,1 eq.) and trimethylamine (1 eq.). The mixture was stirred at room temperature and then concentrated in vacuo to afford ZU5 as a yellow oil with 97% of purity (HPLC). HPLC analysis {BEH C18 type, mobile phase: H20/acetonitrile (HCOOH 0.1%)): tR = 2.44 minPreparation of ZUe and ZU6:l-(3-(9H-xanthen-9-ylidene)propyl)piperidine-4-carboxylic acidand eth -(3-(9H-xanthen-9-ylidene)propyl)piperidine-4-carboxylate
To a solution of 9-oxoxanthene (1.0 equiv.) in THF at reflux were added a solution of cyclopropylmagnesium bromide in THF (1.0 equiv.) and stirred during 2 hours. The mixture was cooled down at room temperature and a solution of hydrogen bromide in acetic acid (4 eq.) was added and stirred at room temperature. The reaction mixture was concentrated in vacuo and purified by silica gel column chromatography to obtain 9- (3bromopropylidene)-oxoxanthene.Then, to a solution of 9-(3bromopropylidene)-oxoxanthene in acetonitrile at reflux was added Ethyl 4-piperidinecarboxylate (1,5 eq.), potassium iodide (0.1 eq.) and potassium carbonate (3 eq.). The mixture was stirred at reflux then concentrated in vacuo and purified by silica gel column chromatography to obtain ZU6 ZU6 is then dissolved in a mixture of THF and a solution of NaOH in water. After phase separation, the aqueous layer was extracted twice by diethyl ether. The global organic layer was, then, washed by a saturated solution of NaCl, dried over MgSC^, filtered and concentrated in vacuo to afford ZUe as a white solid with a purity up to 97% in HPLC. Preparation of ZUc:(Z)-2-(4-(3-(2-(trifluoromethyl)-9H-thioxanthen-9-ylidene)propyl)piperazin-l-yl) ethanamine
EtOH, reflux, 2hPurification withHCI buffer
To a solution of ZU1 (2-[4-[3-[2-(trifiuoromethyl)thioxanthen-9- ylidene]propyl]piperidin-l-yl] ethanol) (leq.) in THF was added diethylazodicarboxylate, phtalimide and triphenylphosphine. The solution was stirred at room temperature during 3 hours and then concentrated in vacuo. The crude oil was then dissolved in ethanol, hydrazine was added and the mixture was stirred at reflux during 2 hours. The crude product obtained after concentration was purified via a reversed phase chromatography using HCI as buffer to afford the compound ZUc as an hydrochloride salt (orange solid). [M+H]+ (ESI+) : 434. HPLC analysis (BEH C18 type, mobile phase: H20/acetonitrile (HCOOH 0.1%)): tR = 2.04 min Preparation of ZUd:(Z)-l-(2-fluoroethyl)-4-(3-(2-(trifluoromethyl)-9H-thioxanthen-9-ylidene)propyl) iperazine
To a solution of flupenthixol in dichloromethane was added at -10°C diethylamino sulfur trifluoride. The mixture was then stirred at room temperature. The crude product was purified via a reversed phase chromatography using HC1 as buffer to afford the compound ZUd as a hydrochloride salt (orange solid) with 97% purity in HPLC. HPLC analysis (BEH C18 type, mobile phase: H20/acetonitrile (HCOOH 0.1%)): tR = 3.59 min.Compounds ZU, ZUa, ZUb, ZU1, ZU2The following compounds can be easily found in commerce:
Example 2: Ebselen oxide derivatives
AS ON DEC2021 3,491,869 VIEWS ON BLOG WORLDREACH AVAILABLEFOR YOUR ADVERTISEMENT
Zuclopenthixol antagonises both dopamine D1 and D2 receptors, α1-adrenoceptors and 5-HT2 receptors with a high affinity, but has no affinity for cholinergic muscarine receptors. It weakly antagonises the histamine (H1) receptor but has no α2-adrenoceptor blocking activity[citation needed].
Evidence from in vitro work and clinical sources (i.e. therapeutic drug monitoring databases) suggests that both CYP2D6 and CYP3A4 play important roles in zuclopenthixol metabolism.[11]
Pharmacokinetics
History
Zuclopenthixol was introduced by Lundbeck in 1978.[22]
^ Parent M, Toussaint C, Gilson H (1983). “Long-term treatment of chronic psychotics with bromperidol decanoate: clinical and pharmacokinetic evaluation”. Current Therapeutic Research. 34 (1): 1–6.
^ Jump up to:ab Jørgensen A, Overø KF (1980). “Clopenthixol and flupenthixol depot preparations in outpatient schizophrenics. III. Serum levels”. Acta Psychiatrica Scandinavica. Supplementum. 279: 41–54. doi:10.1111/j.1600-0447.1980.tb07082.x. PMID6931472.
^ Jump up to:ab Reynolds JE (1993). “Anxiolytic sedatives, hypnotics and neuroleptics.”. Martindale: The Extra Pharmacopoeia (30th ed.). London: Pharmaceutical Press. pp. 364–623.
^ Ereshefsky L, Saklad SR, Jann MW, Davis CM, Richards A, Seidel DR (May 1984). “Future of depot neuroleptic therapy: pharmacokinetic and pharmacodynamic approaches”. The Journal of Clinical Psychiatry. 45 (5 Pt 2): 50–9. PMID6143748.
^ Young D, Ereshefsky L, Saklad SR, Jann MW, Garcia N (1984). Explaining the pharmacokinetics of fluphenazine through computer simulations. (Abstract.). 19th Annual Midyear Clinical Meeting of the American Society of Hospital Pharmacists. Dallas, Texas.
^ Janssen PA, Niemegeers CJ, Schellekens KH, Lenaerts FM, Verbruggen FJ, van Nueten JM, et al. (November 1970). “The pharmacology of fluspirilene (R 6218), a potent, long-acting and injectable neuroleptic drug”. Arzneimittel-Forschung. 20 (11): 1689–98. PMID4992598.
^ Beresford R, Ward A (January 1987). “Haloperidol decanoate. A preliminary review of its pharmacodynamic and pharmacokinetic properties and therapeutic use in psychosis”. Drugs. 33 (1): 31–49. doi:10.2165/00003495-198733010-00002. PMID3545764.
^ Reyntigens AJ, Heykants JJ, Woestenborghs RJ, Gelders YG, Aerts TJ (1982). “Pharmacokinetics of haloperidol decanoate. A 2-year follow-up”. International Pharmacopsychiatry. 17 (4): 238–46. doi:10.1159/000468580. PMID7185768.
^ Larsson M, Axelsson R, Forsman A (1984). “On the pharmacokinetics of perphenazine: a clinical study of perphenazine enanthate and decanoate”. Current Therapeutic Research. 36 (6): 1071–88.
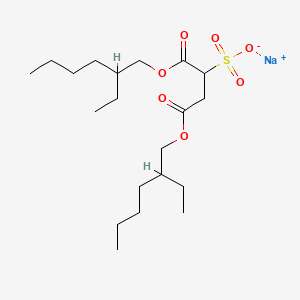
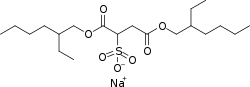
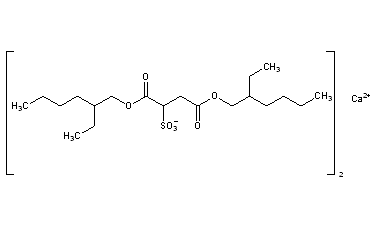
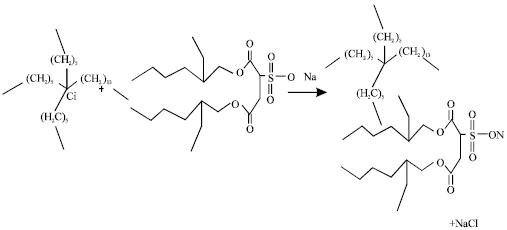




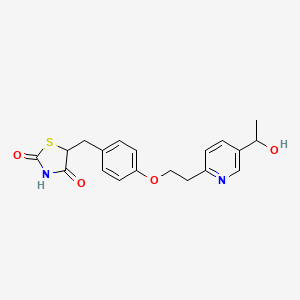
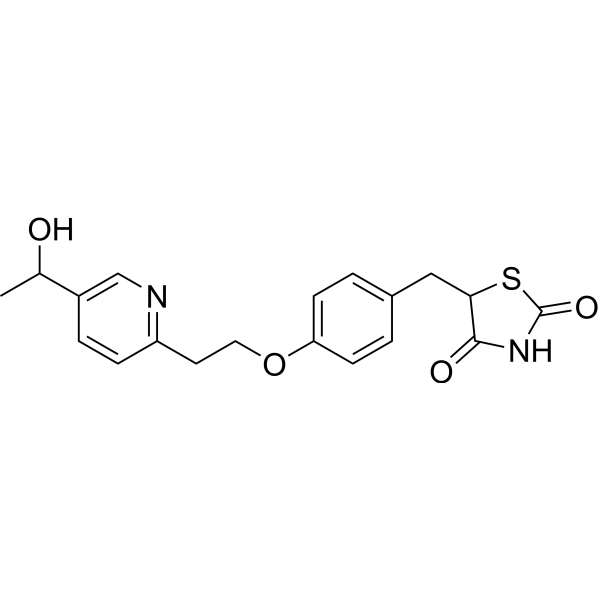
























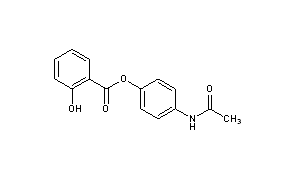





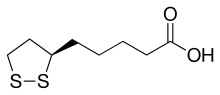


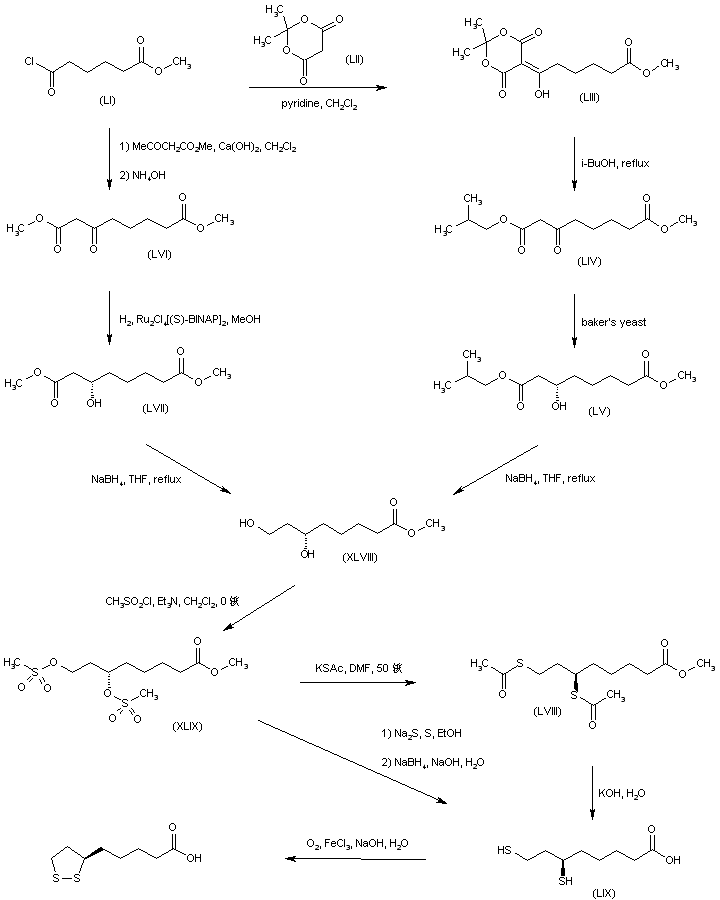
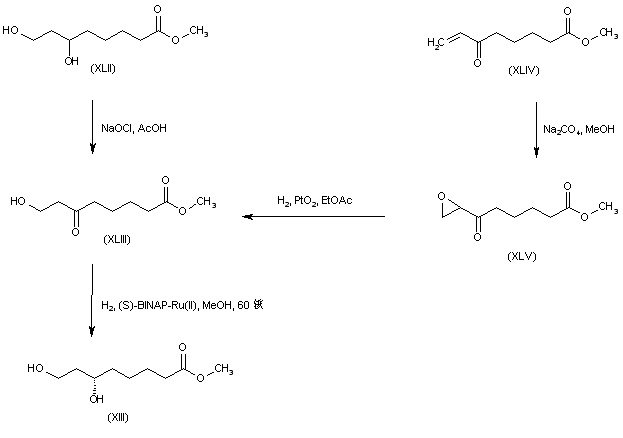
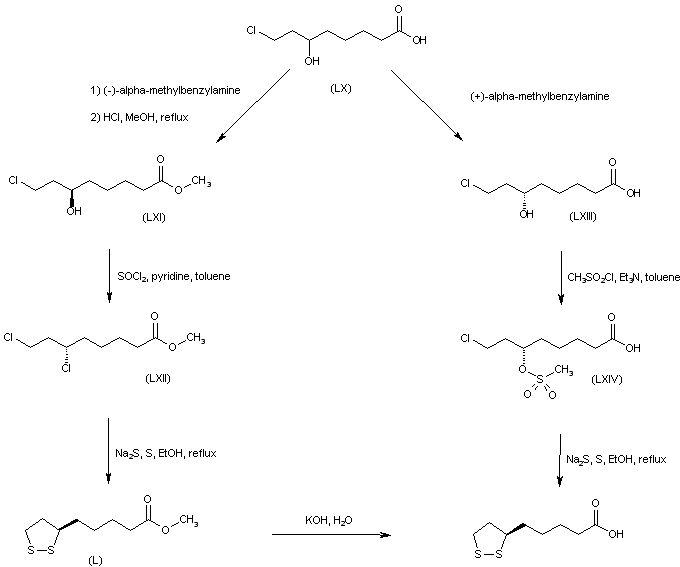
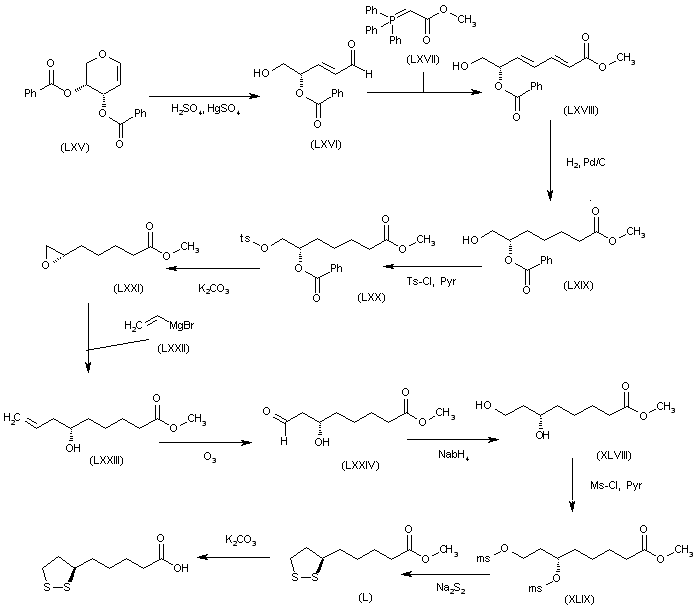
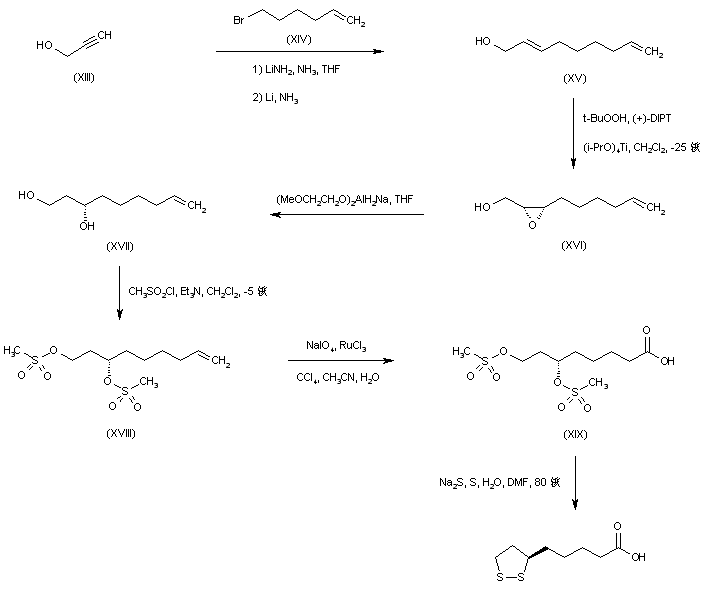
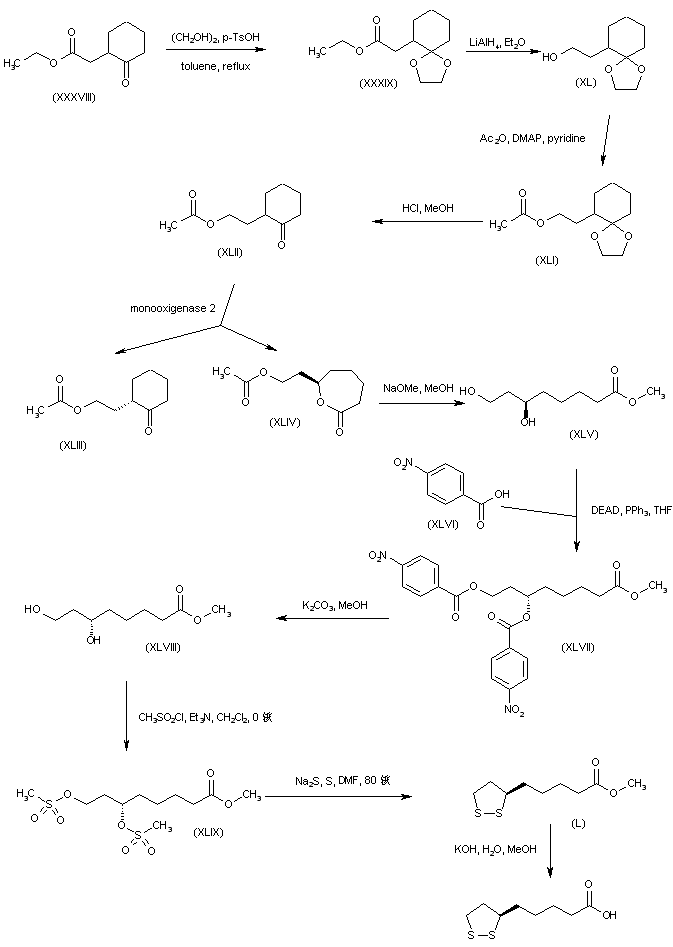
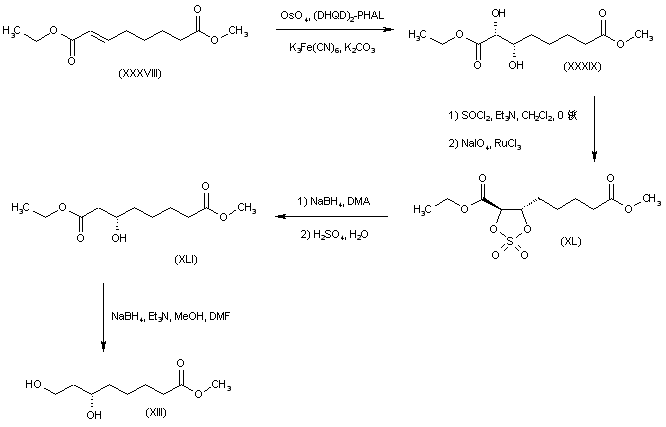
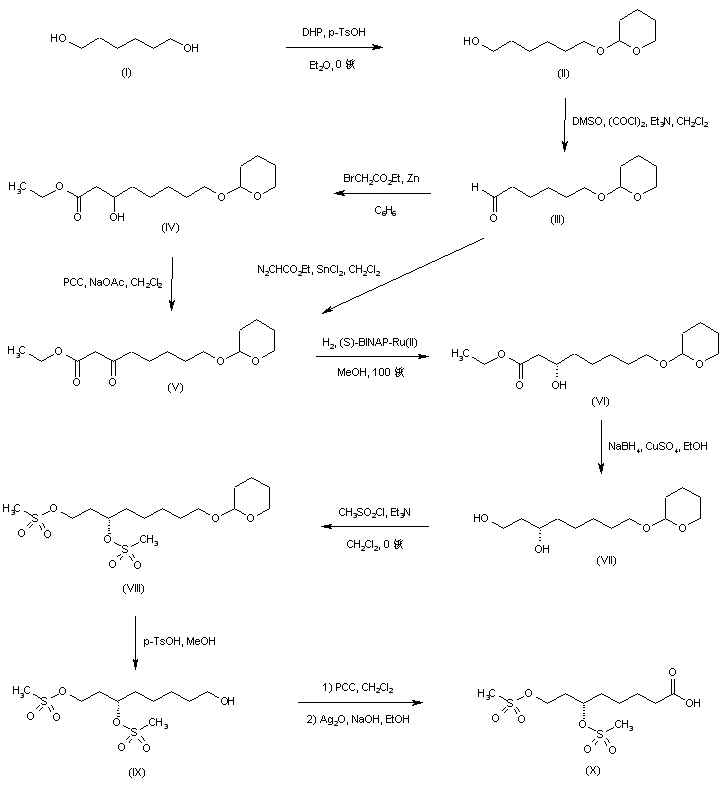
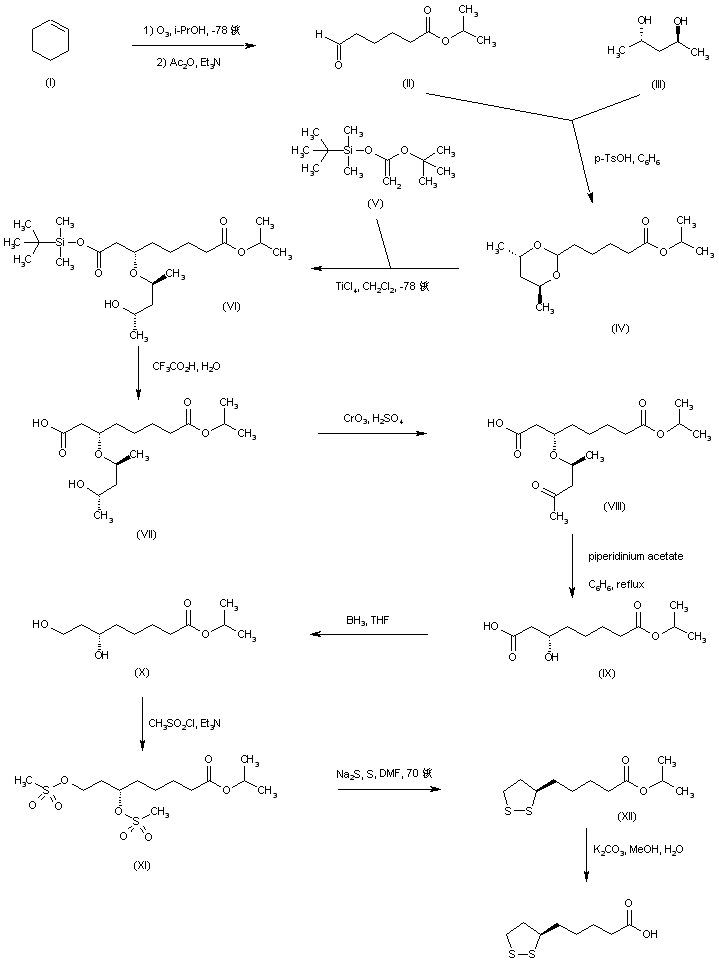
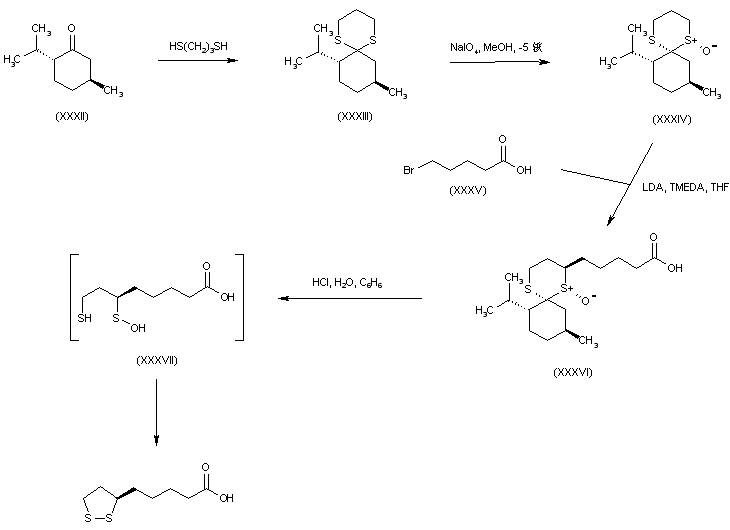
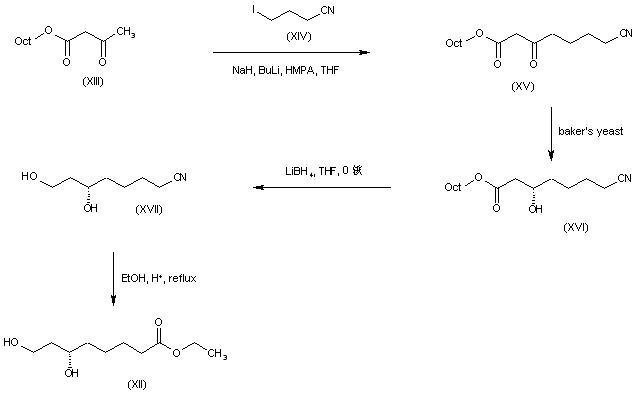
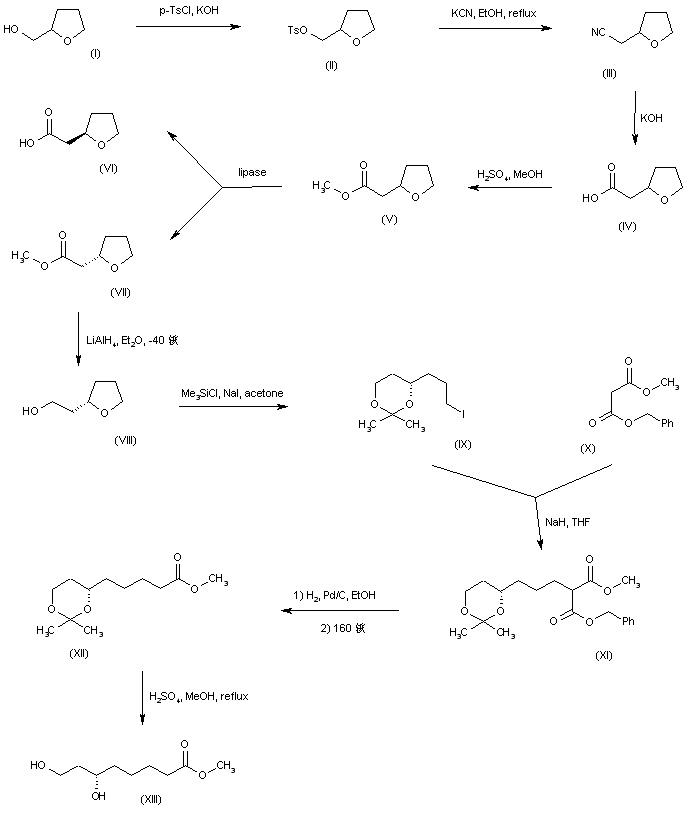
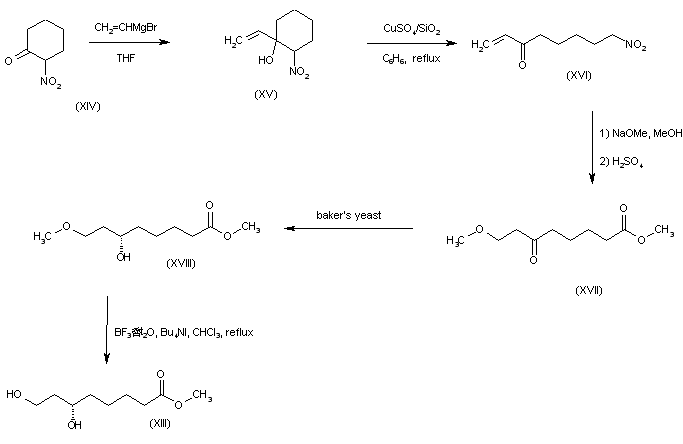
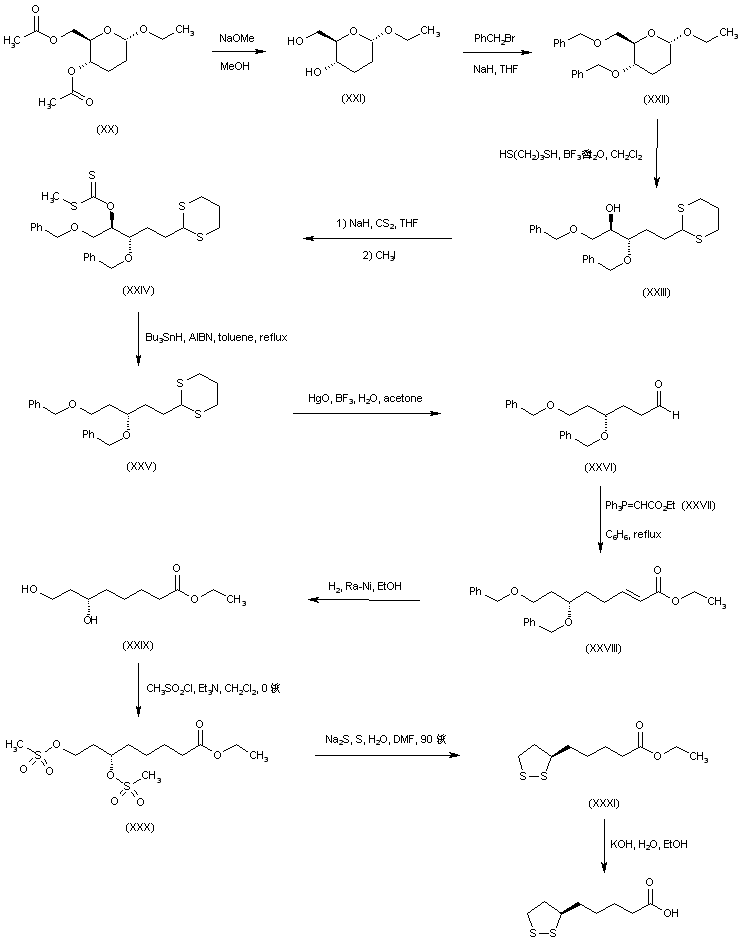



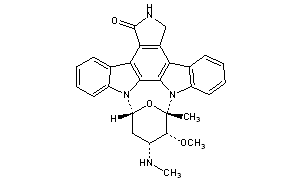


















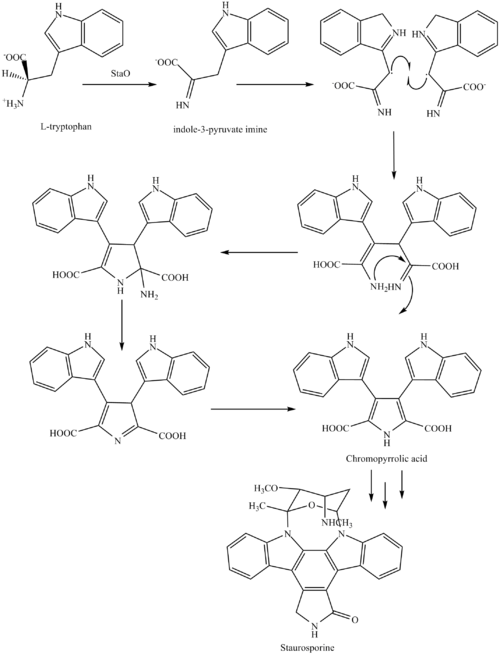










 (Aviptadil-acetate) and Submits Emergency Use Authorization Application to USFDA to Treat Critical COVID-19 in Patients Suffering from Respiratory Failure.
(Aviptadil-acetate) and Submits Emergency Use Authorization Application to USFDA to Treat Critical COVID-19 in Patients Suffering from Respiratory Failure.

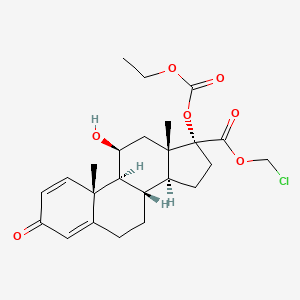





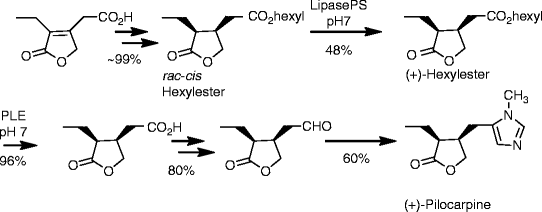






![[(2R,3S,4R,5R)-5-(4-amino-2-oxopyrimidin-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl [hydroxy-[2-(trimethylazaniumyl)ethoxy]phosphoryl] phosphate.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=11583971&t=l)