Immunoglobulin G1, anti-(human transferrin receptor) (human-mus musculus monoclonal JR-141 gamma1-chain) fusion protein with peptide (synthetic 2-amino acid linker) fusion protein with human iduronate-2-sulfatase, disulfide with human-mus musculus mono
Immunoglobulin G1-kappa, anti-(human transferrin receptor 1, tfr1) humanized monoclonal antibody, fused with human iduronate-2-sulfatase, glycoform alfa:
Pabinafusp alfa is under investigation in clinical trial NCT03568175 (A Study of JR-141 in Patients With Mucopolysaccharidosis II).
JCR Pharmaceuticals Announces Approval of IZCARGO® (Pabinafusp Alfa) for Treatment of MPS II (Hunter Syndrome) in Japan
– First Approved Enzyme Replacement Therapy for MPS II to Penetrate Blood-Brain Barrier via Intravenous Administration, Validating JCR’s J-Brain Cargo® Technology –March 23, 2021 07:30 AM Eastern Daylight Time
HYOGO, Japan–(BUSINESS WIRE)–JCR Pharmaceuticals Co., Ltd. (TSE 4552; “JCR”) today announced that the Ministry of Health, Labour and Welfare (MHLW) in Japan has approved IZCARGO® (pabinafusp alfa 10 mL, intravenous drip infusion) for the treatment of mucopolysaccharidosis type II (MPS II, or Hunter syndrome). IZCARGO® (formerly known as JR-141) is a recombinant iduronate-2-sulfatase enzyme replacement therapy (ERT) that relies on J-Brain Cargo®, a proprietary technology developed by JCR, to deliver therapeutics across the blood-brain barrier (BBB). It is the first-ever approved ERT that penetrates the BBB via intravenous administration, a potentially life-changing benefit for individuals with lysosomal storage disorders (LSDs) such as MPS II.
“Subsequent to this approval in Japan, I look forward to further accumulation of clinical evidence for pabinafusp alfa in Brazil, the US and EU”Tweet this
Many patients with MPS II show complications not only in somatic symptoms but also in the central nervous system (CNS), which are often severe, with significant effects on patients’ neurocognitive development, independence, and quality of life. By delivering the enzyme to both the body and the brain, IZCARGO® treats the neurological complications of Hunter syndrome that other available therapies have been unable or inadequate to address so far.
“Approval of IZCARGO® in Japan under SAKIGAKE designation is a key milestone in JCR Pharmaceuticals’ global expansion. It comes on the heels of Fast Track designation from the US FDA, orphan designation from the European Medicines Agency, and the FDA’s acceptance of the JR-141 Investigational New Drug application, enabling JCR to begin our Phase 3 trial in the US,” said Shin Ashida, chairman and president of JCR Pharmaceuticals. “These critical regulatory milestones in Japan, where we have such a strong record of success, and those in the US and Europe, provide important validation of the value of our J-Brain Cargo® technology to deliver therapies across the blood-brain barrier, which we believe is essential to addressing the central nervous system complications of lysosomal storage disorders. We will continue our uncompromising effort to take on the challenge of providing new treatment options for patients with lysosomal storage disorders around the world as soon as possible.”
The MHLW’s approval of IZCARGO® is based on totality of evidence from non-clinical and clinical studies1-4. In a phase 2/3 clinical trial conducted in Japan, all 28 patients experienced significant reductions in heparan sulfate (HS) concentrations in the cerebrospinal fluid (CSF) – a biomarker for effectiveness against CNS symptoms of MPS II – after 52 weeks of treatment, thus meeting the trial’s primary endpoint. IZCARGO® maintained somatic disease control in patients who switched from standard ERT to IZCARGO®. The study also confirmed an improvement in somatic symptoms in participants who had not previously received standard ERT prior to the start of the trial. Additionally, a neurocognitive development assessment demonstrated maintenance or improvement of age-equivalent function in 21 of the 28 patients. There were no reports of serious treatment-related adverse events in the trial, suggestive of a favorable safety and tolerability profile for IZCARGO®.4
“Subsequent to this approval in Japan, I look forward to further accumulation of clinical evidence for pabinafusp alfa in Brazil, the US and EU,” said Dr. Paul Harmatz of University of California – San Francisco (UCSF) Benioff Children’s Hospital Oakland, Oakland, CA, United States. “The availability of an enzyme replacement therapy that crosses the blood-brain barrier is expected to treat both CNS and somatic symptoms associated with this devastating and life-threatening disorder, including developmental and cognitive delays, bone deformities, and abnormal behavior, which have, historically, been unaddressed.”
JCR recently filed an application with the Brazilian Health Surveillance Agency (Agência Nacional de Vigilância Sanitária [ANVISA]) for marketing approval of IZCARGO® for the treatment of patients with MPS II. JCR is also preparing to launch a Phase 3 trial of IZCARGO® in the US, Brazil, the UK, Germany, and France.
About pabinafusp alfa
Pabinafusp alfa (10 mL, intravenous drip infusion) is a recombinant fusion protein of an antibody against the human transferrin receptor and idursulfase, the enzyme that is missing or malfunctioning in subjects with Hunter syndrome. It incorporates J-Brain Cargo®, JCR’s proprietary BBB-penetrating technology, to cross the BBB through transferrin receptor-mediated transcytosis, and its uptake into cells is mediated through the mannose-6-phosphate receptor. This novel mechanism of action is expected to make pabinafusp alfa effective against the CNS symptoms of Hunter syndrome.
In pre-clinical trials, JCR has confirmed both high-affinity binding of pabinafusp alfa to transferrin receptors, and passage across the BBB into neuronal cells, as evidenced by electron microscopy. In addition, JCR has confirmed enzyme uptake in various brain tissues. The company has also confirmed a reduction of substrate accumulation in the CNS and peripheral organs in an animal model of Hunter syndrome.1
In several clinical trials of pabinafusp alfa, JCR obtained evidence of reduced HS concentrations in the CSF, a biomarker for assessing effectiveness against CNS symptoms. The results were consistent with those obtained in pre-clinical studies. Clinical studies have also demonstrated positive effects of pabinafusp alfa on CNS symptoms.2
About J-Brain Cargo® Technology
JCR’s first-in-class proprietary technology, J-Brain Cargo®, enables the development of therapies that cross the BBB and penetrate the CNS. The CNS complications of diseases are often severe, resulting in developmental delays, an impact on cognition and, above all, poor prognosis, which affect patients’ independence as well as the quality of life of patients and their caregivers. With J-Brain Cargo®, JCR seeks to address the unresolved clinical challenges of LSDs by delivering the enzyme to both the body and the brain.
About Mucopolysaccharidosis II (Hunter Syndrome)
Mucopolysaccharidosis II (Hunter syndrome) is an X-linked recessive LSD caused by a deficiency of iduronate-2-sulfatase, an enzyme that breaks down complex carbohydrates called glycosaminoglycans (GAGs, also known as mucopolysaccharides) in the body. Hunter syndrome, which affects an estimated 7,800 individuals worldwide (according to JCR research), gives rise to a wide range of somatic and neurological symptoms. The current standard of care for Hunter syndrome is ERT. CNS symptoms related MPS II have been unmet medical needs so far.
About JCR Pharmaceuticals Co., Ltd.
JCR Pharmaceuticals Co., Ltd. (TSE 4552) is a global specialty pharmaceuticals company that is redefining expectations and expanding possibilities for people with rare and genetic diseases worldwide. We continue to build upon our 45-year legacy in Japan while expanding our global footprint into the US, Europe, and Latin America. We improve patients’ lives by applying our scientific expertise and unique technologies to research, develop, and deliver next-generation therapies. Our approved products in Japan include therapies for the treatment of growth disorder, Fabry disease, acute graft-versus host disease, and renal anemia. Our investigational products in development worldwide are aimed at treating rare diseases including MPS I (Hurler syndrome, Hurler-Scheie, and Scheie syndrome), MPS II (Hunter syndrome), Pompe disease, and more. JCR strives to expand the possibilities for patients while accelerating medical advancement at a global level. Our core values – reliability, confidence, and persistence – benefit all our stakeholders, including employees, partners, and patients. Together we soar. For more information, please visit https://www.jcrpharm.co.jp/en/site/en/.
1 Sonoda H, Morimoto H, Yoden E, et al. A blood-brain-barrier-penetrating anti-human transferrin receptor antibody fusion protein for neuronopathic mucopolysaccharidosis II. Molecular Therapy. 2018;26(5):1366-1374.
2 Morimoto H, Kida K, Yoden E, et al. Clearance of heparan sulfate in the brain prevents neurodegeneration and neurocognitive impairment in MPS II mice. Molecular Therapy. 2021;S1525-0016(21)00027-7.
3 Okuyama T, Eto Y, Sakai N, et al. Iduronate-2-sulfatase with anti-human transferrin receptor antibody for neuropathic mucopolysaccharidosis II: a phase 1/2 trial. Molecular Therapy. 2019;27(2):456-464.
4 Okuyama T, Eto Y, Sakai N, et al. A phase 2/3 trial of pabinafusp alfa, IDS fused with anti-human transferrin receptor antibody, targeting neurodegeneration in MPS-II. Molecular Therapy. 2021;29(2):671-679.
//////////Pabinafusp alfa, JR-141, JR 141,APPROVALS 21, JAPAN 2021
1H-1,2,3-Triazolium, 3-(((2S,3S,5R)-2-carboxy-3-methyl-4,4-dioxido-7-oxo-4-thia-1-azabicyclo(3.2.0)hept-3-yl)methyl)-1-methyl-, inner salt
Enmetazobactam
The Board of directors of Orchid Pharma Ltd has announced that the company had developed a new molecule known as OCID-5090, which was licensed to a company named Allecra Therapeutics, this molecule was undergoing the clinical trials and the company is happy to announce that the molecule has cleared the Phase 3 clinical trials.
Allecra Therapeutics would now either directly or through out license file for NDA of this molecule. Allecra has already out licensed the product to Haini Pharmaceuticals, China for the Chinese Territory at a value of $78mn plus royalties.
As per the IP Agreement between Orchid Pharma Limited and Allecra Therapeutics, Orchid is entitled to receive a Royalty of 6-8% on the worldwide sales of the product. Therefore, once the molecule is commercialised, Orchid can expect a regular stream of Royalty from Allecra. Further, the rights to develop and commercialise the molecule in India (which is under patent protection) remain with Orchid Pharma Limited, and the company is evaluating the various options to commercialise the product.
Orchid had developed a new molecule known as OCID-5090, which was licensed to a company named Allecra Therapeutics, this molecule was undergoing the clinical trials and the molecule has cleared the Phase 3 clinical trials.
Allecra Therapeutics would now either directly or through out license file for NDA of this molecule. Allecra has already out licensed the product to Haini Pharmaceuticals, China for the Chinese Territory at a value of $78mn plus royalties.
As per the IP Agreement between Orchid Pharma Limited and Allecra Therapeutics, Orchid is entitled to receive a Royalty of 6-8% on the worldwide sales of the product. Therefore, once the molecule is commercialised, Orchid can expect a regular stream of Royalty from Allecra. Further, the rights to develop and commercialise the molecule in India (which is under patent protection) remain with Orchid Pharma Limited, and the company is evaluating the various options to commercialise the product.
INVENTOR
Senthilkumar U P
ORCHID
Summary of Profile of Dr. U. P. Senthilkumar, R&D Centre, Orchid Pharma Ltd. Dr. U. P. Senthilkumar Ph.D., the principal inventor of novel beta-lactamase inhibitor, OCID5090, is currently serving as the senior vice-president at Orchid’s Research and Development Centre, Chennai. With illustrious credentials — top ranks in B.Sc. and M.Sc. degrees, first rank in the Graduate Aptitude Test in Engineering (GATE), UGC-CSIR Junior Research Fellowship (JRF) and the prestigious Dr. K.S. Krishnan Fellowship from the Department of Atomic Energy (DAE) and publication of M.Sc. project work in the Indian Journal of Chemistry in 1987 — Mr. Senthilkumar chose to pursue his doctoral research in synthetic organic chemistry with his mentor Prof. Ramasubbu Jeyaraman at Bharathidasan University, Tiruchirapalli. His research focus on the conformational preferences of sterically challenged novel N-Nitroso heterocycles and their conformation dependent anti-cancer properties, led to the publication of 9 articles in reputed peer-reviewed international journals – a commendable accomplishment in the 90s. After a brief post-doctoral stint on fluorescent dicyclopentapyridines, Dr. U. P. Senthilkumar joined Torrent Research Centre at Ahmedabad and started his new endeavor of drug discovery on ACE inhibitors. At the process research and development laboratory, he was actively involved in asymmetric and stereo-selective synthesis of Active Pharmaceutical Ingredients (APIs), and exploited the full potential of chiral prep-HPLC to realize the target molecules. After joining Orchid Pharma Ltd., Chennai, Dr. Senthilkumar led the efforts in the development of differentiated and patentable manufacturing processes for APIs related to both non-antibiotics and beta-lactam antibiotics. He played a significant role in successfully implementing the manufacturing processes overcoming several challenging problems. In addition, his scientific insights and breath of understanding on the patent landscape were invaluable and impactful in creating significant value to the organization and growth of the company in realizing the mission to become a leader in the pharmaceutical generic business. One finds more than 100 articles/patents/publications to his credit, which include inventions on new drugs, drug-intermediates, products, processes, new synthetic routes, rearrangements and novel polymorphs. As a Leadership Persona of the IP management team, he had exhibited a thoroughness of the science/invention and meticulously executed the task of prosecution of few hundred patents in many countries from both New Drug Discovery and Process Chemistry space. All the successful effort earned Orchid Pharma Ltd the National Intellectual Property Award from the Department of Industrial Policy and Promotion, Ministry of Commerce and Industry,Government of India. Through his executive and decision-making skills combined with scientific rationale and clarity, Dr. Senthilkumar played significant role in the selection of products and creation of generic product portfolio for Orchid, with unique IP strategies, analysis of patents, patent mapping, designing & developing invalidation/non-infringing positions, and early launch opportunity, including first-to-file (FTF) positioning. His appearance in the US courts, for deposition in couple of patent litigations, and successful accomplishment of the same are testimony to his depth, thoroughness of science and the ability to defend the invention with grit and professionalism. Additionally, his effectual role in the first-to-launch of one of the large volume sterile penicillins with regulatory exclusivity, achieved successfully by overcoming the citizen petition process in the regulatory pathway, is another shining example of his leadership and scientific strength. To support in-house projects as well as multinational pharma majors, Dr. Senthilkumar has taken up CRAMS (Contract Research and Manufacturing Services) and CMC (Chemistry, Manufacturing and Control) for new chemical entities. Besides, he passionately focused on novel beta-lactamase inhibitors and their antibiotic combinations that were envisaged by him to exhibit potent activity against multi-drug resistant bacteria. His dedicated effort brought a novel extended spectrum beta-lactamase inhibitor, OCID5090, which was out-licensed to Allecra Inc. OCID5090/cefepime combination has completed successfully the Phase III clinical trials for treating complicated urinary-track-infections (cUTI), including acute pyelonephritis (AP), and rightfully, OCID5090 has gotten the US FDA fast track designation as a Qualified Infectious Disease product (QIDP) that provides a five-year additional market exclusivity and priority review. His never-ending passion for research is infectious and roped him with academic institutions to explore novel technologies including electron-beam irradiated heterogeneous catalysis. His commendable knowledge on intellectual property is being utilized by the IP Cells of various institutions as well as the Tamil Nadu State Technology Development and Promotion Centre. A sincere student he is, Dr. Senthilkumar is also a founder-member of Prof. Ramasubbu Jeyaraman Science Foundation (RJSF). Since 2011, he has been playing a significant role in rganizing several academic events (seminars, work-shops, invited lectures, state-level proficiency tests, and research-orientation programs) for post-graduate chemistry students to create passion for research. His concern and help for poor and rural students show his human face.
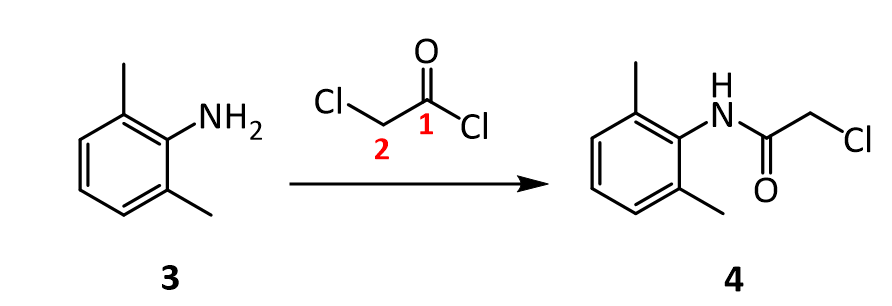
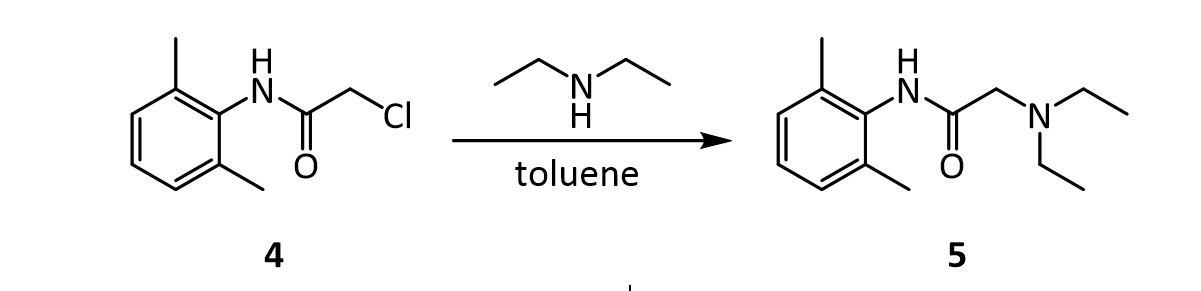
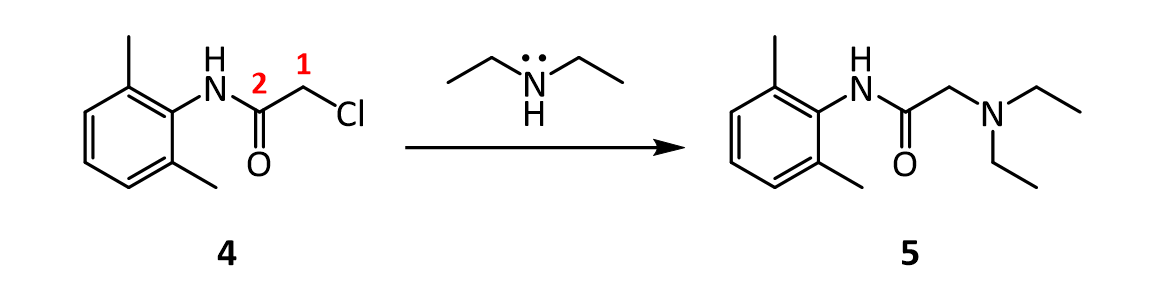
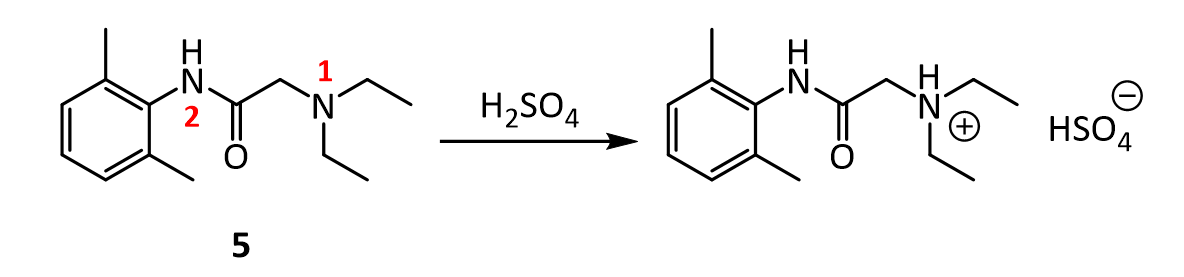
[0051]To a suspension of (2S,3S,5R)-3-methyl-7-oxo-3-(1H-1,2,3-triazol-1-ylmethyl)-4-thia-1-azabicyclo-[3.2.0]heptane-2-carboxylic acid 4,4-dioxide (25 g) in acetone (100 mL) at 25-30° C. was added slowly N,O-bis(silylacetamide) (18.6 g) with stirring. The reaction mixture was stirred at this temperature (25-30° C.) for 15-20 min. To the clear solution obtained, methyl iodide (100 mL) was added over a period of 15 min. and stirred at 25-30 min. for 24 h. The precipitated solid was separated by filtration and washed with acetone (25 mL). Wet weight of the solid obtained was 30 g.
[0052]The above wet solid was stirred with purified water (300 mL) at 10-15° C. for 2.5 h. To the resulted reaction mixture was added sodium thiosulfate (0.1 g) and stirred at 10-15° C. for 10-15 min. To the reaction mixture, dichloromethane (300 mL) was added, stirred and the organic layer separated. The aqueous layer was washed with a solution of Amberlite LA-2 resin (5% solution in dichloromethane twice, followed by dichloromethane twice. To the aqueous solution, activated carbon (1 g) was added, stirred for 15 min, filtered and washed with purified water (25 mL). The solution was filtered and lyophilized to get the title compound in pure form (10 g). 1H NMR (400 MHz, DMSO) δ ppm: 1.39 (s, 3H), 3.14 (dd, J=16.0, 1.3 Hz, 1H), 3.55 (dd, J=16.0, 4.2 Hz, 1H), 3.97 (s, 1H), 4.34 (s, 3H), 5.05 (dd, J=4.2, 1.3 Hz, 1H), 5.29 (d, J=14.7 Hz, 1H), 5.42 (d, J=14.7 Hz, 1H), 8.91 (d, J=1.3 Hz, 1H), 8.99 (d, J=1.3 Hz, 1H). Mass m/z: M+1 peak at 315. Alternatively the solution could be subjected to spray-drying to yield the title compound.
Synthesis of (2535.5R)-3-methyl-3-((3-methyl-lH-1.2 -triazol-3-ium-l-yl)methvn-7-oxo-4-thia-l-azabicyclor3.2.01heptane-2-carboxylate 4,4-dioxide (4),
Compound (4) was prepared according to Scheme 2.
Scheme 2
i) Ν,Ο-bis-trimethylsilylacetamide, CH2CI2; ii) CH3OTf; iii) Na 2-ethylhexanoate
In a round bottom flask under nitrogen flow 100 g of Tazobactam acid (1) and 500 mL of Dichloromethane are loaded. The temperature is adjusted to +30/35°C then 37 g of Ν,Ο-Bis(trimethylsilyl) acetamide are loaded in 15-20 minutes maintaining the temperature to +35/42°C. The mixture is heated to reflux (+40/42°C) for 60 minutes. If the solution is not clear, N,0-Bis(trimethylsilyl) acetamide is loaded in small portions (0,5-1.0 g each) waiting 15 minutes every time till a clear solution containing intermediate (2) is obtained. 0.55 moles of N,0-Bis(trimethylsilyl) acetamide is used, with further 0.1-0.2 equivalents being added if the reaction is not complete.
Then the temperature is cooled down to 0/+5°C and 70 g of Methyl trifluoromethanesulfonate are loaded in 60-90 minutes maintaining the temperature at 0/+5°C. After 30 minutes the reaction is monitored by HPLC to control the disappearance of intermediate (2) and formation of intermediate (3). The reaction is monitored every 30 minutes until completion.
In a round bottom flask, under nitrogen, are loaded 500 mL of Ethanol and 55 g of Sodium 2-Ethylhexanoate and the temperature is adjusted to +20/25°C, then the reaction solution containing intermediate (3) is added in 60-90 minutes maintaining the temperature of +20/25 °C under vigorous stirring. The suspension is stirred for 30 minutes then is filtered and washed with 300 mL of Ethanol followed by 500 mL of Dichloromethane under nitrogen. The crude product (4) is dried under nitrogen flow till constant weight (150 g) is obtained. The crude product compound (4) was isolated as a solid product (HPLC assay = 70%, yield = 80%).
Purification of (2tS’,3^5^)-3-methyl-3-((3-methyl-lH-l,2,3-triazol-3-ium-l-yl)methyl)-7-oxo-4-thia-l-azabicyclor3.2.01heptane-2-carboxylate 4,4-dioxide (4)
In a round bottom flask 800 mL of Dimethylformamide are loaded, the temperature is adjusted to +20/25°C then crude Compound 4 (150g) obtained above is loaded using 100 mL of Dimethylformamide to facilitate the transfer. The mixture is stirred for 5 minutes and a solution is obtained, then and after a few minutes crystallization takes place. The suspension is stirred for about 3 hours, then is cooled to 0/+5°C and stirred for another 3 hours.
The solid is filtered and washed with 300 mL of Dimethylformamide pre-cooled to 0/+5°C. Compound 4 is then suspended in 700 mL of Ethyl acetate and the temperature is adjusted to +40/45°C. The suspension is stirred for 30 minutes then the solid is filtered and washed with 150 mL of Ethyl acetate pre-heated to +40/45°C. The suspension with
Ethyl acetate is repeated twice. Finally Compound 4 is dried under vacuum at +40°C till constant weight is achieved (66 g, HPLC assay = 99%, yield = 76%).
Compound 4 Sterile filtration and recrystallization Procedure
In a round bottom flask 350 mL of Methanol are loaded, the temperature is adjusted to +30/35°C then 100 g of Compound 4 are loaded and finally the flask is washed with 60 mL of Methanol. After 5-10 minutes a solution is obtained. The solution is diluted with 330 mL of acetone adjusting the temperature to +20/+25°C. The obtained solution is treated with 2,2 g of charcoal for 20 minutes then filtered using a 0.22microM filter and the filter is washed with a mixture of 13 mL of Methanol and 110 mL of Acetone. The temperature of the solution is adjusted to +30/35°C and under vigorous stirring 830 mL of Acetone are loaded in about 15-20 minutes. After stirring for 60 minutes at temperature of +30/35°C 1170 mL of Acetone are loaded in 45-60 minutes. Then the temperature is adjusted to +20/25 °C in about 30-60 minutes and maintained for 30 minutes. The obtained crystalline solid is filtered and washed with 430 mL of Acetone. Finally the product is dried under vacuum at +40°C till constant weight is achieved (83 g of Compound 4) are obtained with an HPLC assay = 98-99%, yield =t 80%).
Mr. Ram Gopal Agarwal
Chairman and Non-Executive Director
Mr. Ram Gopal Agarwal is Founder Chairman of Dhanuka Group.
He is a decisive and action oriented visionary who took over a sick pesticide Company named Northern Mineral Pvt. Ltd. in 1980 and transformed it today into a Rs 1000 Crore organization called Dhanuka Agritech Ltd.
His deep commitment and inspiring leadership in initial turbulent days is an example worth inculcating and his passion to contribute to Indian Agriculture is commendable.
His ability to prioritize and deal effectively with a number of tasks simultaneously reinforced with the skills to make effective decisions, has metamorphosed the business venture into one of the fastest growing Agrochemical Company in India which has thrice been rated as ‘Best under a Billion Company’ by Forbes Magazine.
In order to achieve his aspiration of “Transforming India through Agriculture” he has dedicated himself to bring changes in Agrochemicals Industry and the farming community. His contribution for adopting newer farming techniques at the grass root level, judicious use of agro chemicals in farming and imparting knowledge through his nationwide network of distributors and Dhanuka Doctors in field has resulted in the overall prosperity of farmers.
Mr. Ram Gopal Agarwal has been the past Chairman of CCFI, (Crop Care Federation of India) the apex Chamber of all Indian Agrochemical majors. He is also Chairman of Advisory Committee of AGRO Chemicals Federation of India.
Mr. Ram Gopal Agarwal, Group Chairman, has been bestowed with many Awards for his tremendous contribution in Agro Industry like “Life Time Achievement Award” by Agri Business Summit and Agri Awards 2019, “Distinguished Contribution to Indian Agrochemicals Industry” during India Chem 2016 International Conference organised by FICCI etc.
Mr. Manish Dhanuka
Managing Director
Mr. Manish Dhanuka is the Director of Orchid Pharma Limited; he has the vision to rejuvenate Orchid Pharma Ltd. and take it on a fruitful path. His wide-ranging experience of handling operations, commercial, marketing and finance in the manufacturing industry provides for his analytical and decision-making skills facilitating the restoration of the company to its glorious past and to achieve even greater heights.
He excels in creating economical Pharmaceutical technologies and accelerated evaluation process for improving healthcare. Experience of 25 years in research, evaluation, and teaching in the pharmaceutical industry equips him with the expertise in innovative pharmaceutical technologies…
He holds a B.Tech in Chemical Engineering from IIT, New Delhi, and M.S in Chemical Engineering from the University of Akron, USA.
Before establishing Dhanuka Laboratories Ltd. in 1993, he began his career at Ranbaxy Labs Ltd. in New Delhi and worked there for 5 years. His vision and strategy to grow the Pharmaceutical industry in the Indian sub-continent, have helped the Dhanuka Group of companies enhance its Bulk Drugs manufacturing arm exponentially. He spearheaded the acquisition of Synmedic Laboratories in the year 2013 which is involved in pharmaceutical formulations. This entrepreneurial vigor enabled him to take over the operations of Orchid Pharma Ltd. in March 2020.
Outside of work, he likes to travel for wildlife adventures.
Mr. Mridul Dhanuka
Whole-Time Director
He is associated with Dhanuka Group Ltd. since 2005. He was responsible in successfully realigning the entire supply chain vertical from procurement to sales. At Orchid, he hopes to replicate the Group’s success and put another feather in Dhanuka cap.
CLIP
Orchid Chemicals & Pharmaceuticals, or Orchid Pharma since its recent name change in 2015, was established in 1992 in Chennai to manufacture antibiotics, and entered drug discovery in 2001 with projects in the areas of anti-infectives and treatments for pain.32, 197 In 2002, the company engaged in a joint venture to develop US-based firm Bexel Biotechnology’s BLX-1002, an oral, non-PPAR AMPK activator for the treatment of diabetes,198 later repositioned for NASH (2012), but no further progress has been reported recently.197 In 2008, Orchid invested in Diakron Pharmaceuticals, a US-based company that had an exclusive license to MSD′s investigational oral anticoagulant drug, a direct thrombin inhibitor later known as DPOC-4088 (or DP-4088),199 which reached Phase 1 clinical studies in Europe in 2012 (Supporting Information Table 6b, entries 5–6).200 The company’s own internal discovery efforts had a broad therapeutic focus, covering infectious diseases, inflammation, pain, oncology, metabolic disorders, and CNS diseases. OCID-2987,197, 201 a PDE4 inhibitor for the treatment of inflammatory disorders such as COPD, completed successfully Phase 1 studies in Europe in 2012, and OCID-4681 29,202, 203 a histone deacetylase (HDAC) inhibitor for cancer had received approval in 2011 for Phase 1 studies for solid tumors in India, but we assume both have been abandoned, as cancer and inflammation are not mentioned in the company’s latest annual reports.197 Two additional compounds were abandoned at the preclinical stage: OCID-5005, a STAT-3/IL-6 inhibitor for oncology, and a unnamed Th1/Th2 cytokine synthesis inhibitor for inflammation (Supporting Information Table 2a, entries 134–138).197 Financial issues led Orchid, as of 2009, to sell parts of its business to Hospira (now part of Pfizer). As a consequence, no progress has been reported on its discovery programs since 2010, and no further NCE patent application has been published since 2012. However, in 2013 Orchid licensed its broad-spectrum β-lactamase inhibitor OCID-5090, a zwitterionic N-methylated tazobactam derivative, to the German Allecra Therapeutics for a 20 % stake in the company, for use in combination with antibiotics to treat multidrug-resistant gram negative bacteria.204–207 Allecra’s lead compound AAI202, a combination of cefepime and AAI101/OCID-5090 30, is currently in Phase 1 studies in France.208, 209
Dr. Gopalan is a synthetic organic chemist with extensive experience in the field of drug discovery and development. After completing his PhD from University of Madras, he went to Harvard University where he worked with the Nobel Laureate, Prof. E.J. Corey, as a post-doctoral fellow. Subsequent to this he joined Syntex Research Inc. in California to work on the synthesis of unnatural amino acids. After a year, he moved to Bristol-Meyers Squibb, Princeton, New Jersey, to contribute to their program on novel antibiotics and ACE inhibitors. Dr. Gopalan then moved back to India in 1982 to join the Drug Discovery Research Division of Boots Pharmaceuticals (India) Ltd. in Mumbai. Over his decade long stint there he contributed extensively to their drug discovery program, and one of the product candidates that he developed went up to Phase-2 clinical trials in both USA and UK. He then moved to Sun Pharma Advanced Research Center as Vice-President and, after a year, took up the position as General Manager at Glaxo (India) Ltd. in 1993. Here, he worked in a broad range of areas that included process development, synthesis of impurities of APIs, and generation of small molecule libraries to support drug discovery efforts to Glaxo, France. In 1999 he took over as Senior Vice President of the Drug Discovery Chemistry Division of Glenmark Pharmaceuticals Ltd. where he was involved in the design and development of inhibitors for PDE IV and DPP IV, as well as agonists for CB2. After a 6-year stint at Glenmark, Dr. Gopalan joined Matrix Laboratories Ltd. as CSO and Executive Vice-President, where he successfully helped to develop novel and selective inhibitors for PDE4 and DPP4. Five years later he became CSO and Executive Director of Orchid Pharmaceuticals Ltd in Chennai. He served in this capacity for close to a decade, contributing extensively to drug design and development in the broad segments of oncology, anti-infectives, and anti-inflammatory & metabolic disorders. Since 2017, Dr. Gopalan has been associated with CSIR-Indian Institute of Chemical Technology as a Scientific Advisor.
Dr. Gopalan’s illustrious career is endowed with numerous successes. He has been inventor, or co-inventor, of several drugs or candidate drugs. These include the novel potassium channel blockers BTS-67582 (BTI-2927) for tpe-2 diabetes, the PDE IV inhibitors Oglemilast (COPD) and Revamilast (RA); DPP IV inhibitor Melogliptin; a selective Cannaboid-2 agonist Tedalinib (Neuropathic pain); a Beta lactamase inhibitor Enmetazobactum (OCID-5090); OCID-18034 (an inhibitor of KPC enzyme); and OCID-18174 (an inhibitor of P. arugenosa). Most of these compounds were out-licensed to major international pharmaceutical companies such as Forest Laboratories Inc. USA, Teijin of Japan, Merck KGaA of Germany, Allecra of Switzerland, and Merck & Co. USA. Dr.Gopalan has 34 publications in National and International Journals, has contributed a Chapter,Co-authored with Professor K.K.Balasubramanian (IITM) on Applications of Click Chemistry in Drug Discovery and Development in a Book on Click reaction in Organic Synthesis, published by Wiley-VCH VERLAG GmbH &Co,KGaA, Weinheim,Germany,Chapter 2, p 25-70,2016, edited by Prof. S. Chandrasekharan (IISc,Bangalore) & 51 Patents.
Commensurate with his achievements, Dr. Gopalan has also received many awards. The more prominent of these include Inventor’s award by Glenmark (2004), Ranbaxy Science Foundation Award in Pharmaceutical Sciences (2005), and the Lifetime Achievement Award in the Field of Chemistry from Vels University (2011).
On May 21, 2021, the Food and Drug Administration granted accelerated approval to amivantamab-vmjw (Rybrevant, Janssen Biotech, Inc.), a bispecific antibody directed against epidermal growth factor (EGF) and MET receptors, for adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.
FDA also approved the Guardant360® CDx (Guardant Health, Inc.) as a companion diagnostic for amivantamab-vmjw.
Approval was based on CHRYSALIS, a multicenter, non-randomized, open label, multicohort clinical trial (NCT02609776) which included patients with locally advanced or metastatic NSCLC with EGFR exon 20 insertion mutations. Efficacy was evaluated in 81 patients with advanced NSCLC with EGFR exon 20 insertion mutations whose disease had progressed on or after platinum-based chemotherapy. Patients received amivantamab-vmjw once weekly for 4 weeks, then every 2 weeks thereafter until disease progression or unacceptable toxicity.
The main efficacy outcome measures were overall response rate (ORR) according to RECIST 1.1 as evaluated by blinded independent central review (BICR) and response duration. The ORR was 40% (95% CI: 29%, 51%) with a median response duration of 11.1 months (95% CI: 6.9, not evaluable).
The most common adverse reactions (≥ 20%) were rash, infusion-related reactions, paronychia, musculoskeletal pain, dyspnea, nausea, fatigue, edema, stomatitis, cough, constipation, and vomiting.
The recommended dose of amivantamab-vmjw is 1050 mg for patients with baseline body weight < 80 kg, and 1400 mg for those with body weight ≥ 80 kg, administered weekly for 4 weeks, then every 2 weeks thereafter until disease progression or unacceptable toxicity.
This indication is approved under accelerated approval based on overall response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
This review was conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence. Project Orbis provides a framework for concurrent submission and review of oncology drugs among international partners. For this review, FDA collaborated with the Brazilian Health Regulatory Agency (ANVISA) and United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA). The application reviews are ongoing at the other regulatory agencies.
This review used the Assessment Aid, a voluntary submission from the applicant to facilitate the FDA’s assessment. The FDA approved this application 2 months ahead of the FDA goal date.
The most common side effects include rash, infusion-related reactions, skin infections around the fingernails or toenails, muscle and joint pain, shortness of breath, nausea, fatigue, swelling in the lower legs or hands or face, sores in the mouth, cough, constipation, vomiting and changes in certain blood tests.[2][3]
Amivantamab was approved for medical use in the United States in May 2021.[2][3][4][5]
Amivantamab, also known as JNJ-61186372, is an anti-EGFR-MET bispecific antibody, derived from Chinese hamster ovary cells, approved for the treatment of adult patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.1,9 Patients with NSCLC often develop resistance to drugs that target EGFR and MET individually, so amivantamab was developed to attack both targets, reducing the chance of resistance developing.1,2 Amivantamab was found to be more effective than the EGFR inhibitor erlotinib or the MET inhibitor crizotinibin vivo.1,3 Patients with NSCLC with exon 20 insertion mutations in EGFR do not respond to tyrosine kinase inhibitors, and were generally treated with platinum-based therapy.5
Amivantamab was granted FDA approval on 21 May 2021.9
Medical uses
Amivantamab is indicated for the treatment of adults with locally advanced or metastatic non-small cell lung cancer (NSCLC) with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, as detected by an FDA-approved test, whose disease has progressed on or after platinum-based chemotherapy.[3]
History
The U.S. Food and Drug Administration (FDA) approved amivantamab based on CHRYSALIS, a multicenter, non-randomized, open label, multicohort clinical trial (NCT02609776) which included participants with locally advanced or metastatic non-small cell lung cancer (NSCLC) with EGFR exon 20 insertion mutations.[3] Efficacy was evaluated in 81 participants with advanced NSCLC with EGFR exon 20 insertion mutations whose disease had progressed on or after platinum-based chemotherapy.[3]
The FDA collaborated on the review of amivantamab with the Brazilian Health Regulatory Agency (ANVISA) and the United Kingdom’s Medicines and Healthcare products Regulatory Agency (MHRA).[3] The application reviews are ongoing at the other regulatory agencies.[3]
Society and culture
Legal status
Amivantamab was approved for medical use in the United States in May 2021.[2][3][4][5] A marketing authorization application is pending in the EU.[6][7]
Amivantamab is being investigated in combination with lazertinib versus osimertinib; and in combination with carboplatin-pemetrexed chemotherapy compared to carboplatin-pemetrexed.[9][10]
). The MET parental mAbs had the F405L mutation and the EGFR parental mAbs had the K409R mutation. The IgG1 b12 arm served as isotype control and null arm to preserve the BsAb architecture. The low fucose parental mAbs were generated using proprietary cell lines. The quality of the BsAb were confirmed as being monodisperse and monomeric via size exclusion chromatography and being pure via SDS-PAGE.
Flow cytometric binding assay
Binding to cells expressing EGFR and MET (A549 [ATCC CCL-185], NCI-H1975 [ATCC, CRL-5908], and NCI-H441 [ATCC HTB-174] cells) was evaluated using flow cytometry (fluorescence-activated cell sorting [FACS]). All BsAbs and controls were diluted in FACS buffer (PBS supplemented with 1% bovine serum albumin and 0.2% sodium azide). After 1 h incubation, unbound antibodies were removed by a FACS buffer wash. The cells were then incubated with goat anti-human IgG-PE (Jackson) for FACS detection (BD FACS Canto). The mean fluorescence intensity of the cells in the live gate was plotted against antibody concentration, and the EC50 was determined by nonlinear regression fitting. Anti-EGFR zalutumumab and anti-MET 5D5 (onartuzumab) were positive controls and anti-CD20 7D8 (Genmab) was the negative control.
MET phosphorylation assay
A549 cells were incubated with 30 μg/ml of test antibody for 15 min and tested for MET phosphorylation using rabbit anti-phospho MET (Tyr1234–1235) (Cell Signaling 3129) and total MET protein using mouse anti-human MET antibody (Cell Signaling 3127). A score of 1 to 4 was given, where 1 = no visible band, 2 = slightly visible band, 3 = phosphorylation comparable with weak agonist (MET B IgG1), and 4 = phosphorylation level similar to positive controls (MET A and MET 5D5 IgG1 mAbs).
Proliferation assays
Test molecules were added to H1975, KP4 (Riken Cell bank, RCB1005), or NCI-H441 cells plated at 5000 or 10,000 (KP4) cells/well in 96-well plates. After 6 (KP4) or 7 (H1975 and NCI-H441) days of incubation at 37 °C and 5% CO2, the number of viable cells was determined using an AlamarBlue assay (Biosource DAL1100). A615 values were measured and plotted in a bar diagram.
EGFR phosphorylation assay
Approximately 106 A549 or SNU-5 cells/well were grown overnight in six-well plates and incubated for 15 min with 30 μg/ml of antibody in the absence or presence of 40 ng/ml EGF. After cell lysis, Western blots determined EGFR phosphorylation status with phospho-EGFR (Tyr1068) antibody (Cell Signaling 2234) and total EGFR protein using an anti-EGFR antibody (Cell Signaling 2232).
Expression and purification of proteins for crystallization
Human MET Sema-PSI region (residues 39–564) containing a C-terminal 8xHis tag was expressed in Tni PRO insect cells infected with recombinant baculovirus. The culture was harvested 72 h post infection, and the MET Sema-PSI protein was purified by affinity and size exclusion chromatography. Briefly, MET was captured with a Ni-NTA resin (Novagen) equilibrated in TBS, 10 mM imidazole, pH 7.4 and eluted from the column with 250 mM imidazole, TBS, pH 7.4. Fractions containing MET were identified by SDS-PAGE and loaded into a Superdex 200 column (GE Healthcare) equilibrated in 20 mM Tris, 50 mM NaCl, pH 7. The final protein concentration was determined by absorbance at 280 nm.The anti-MET Fab of amivantamab was transiently expressed in Expi293F cells. Briefly, the cells were cotransfected with separate plasmids encoding the Fab heavy and light chains at 3:1 (light:heavy chain) molar ratio following transfection kit instructions (Life Technologies). The culture was harvested 5 days post transfection, and the Fab was purified by affinity and cation exchange chromatography. Briefly, the Fab was captured with a HiTrap resin (GE Healthcare) equilibrated in PBS pH 7.2 and eluted from the column with a gradient of 30 to 300 mM imidazole in PBS pH 7.2. The eluate was buffer exchanged into 25 mM NaCl, 20 mM MES pH 6.0, bound to a Source 15S column (GE Healthcare), and eluted with a NaCl gradient in 20 mM MES pH 6.0.
Crystallization and structure determination
The amivantamab anti-MET Fab–MET Sema-PSI complex was prepared by overnight mixing of MET and Fab at a molar ratio of 1:1.3 (excess Fab) at 4 °C, while buffer exchanging to 20 mM Hepes pH 7.0. The complex was captured with a monoS 5/50 column (GE Healthcare) equilibrated in 20 mM Hepes pH 7.0 and eluted from the column with a gradient of NaCl. The complex was concentrated to 4.8 mg/ml.Crystallization trials for the Fab–MET complex were carried out with a Mosquito LCP robot (TTP LabTech) for the setup of sitting drops on 96-well plates (Corning 3550) and a Rock Imager 54 (Formulatrix) for plate storage at 20 °C and automated imaging of drops. Small crystals were initially obtained from 2 M NH4(SO4)2, 0.1 M MES pH 6.5, and they were used as seeds in next rounds of optimization. Crystals suitable for X-ray diffraction were obtained from 2.5 M sodium formate, 5% PEG 400 Da, 0.1 M Tris pH 8.5 after multiple rounds of seeding. The crystals were soaked for a few seconds in a cryoprotectant solution containing mother liquor supplemented with 20% glycerol and then flash frozen in liquid nitrogen. X-ray diffraction data were collected with a Pilatus 6M detector on beamline 17-ID at the Advanced Photon Source (Argonne National Laboratory), and the diffraction data were processed with the program HKL2000. The crystal structure of the Fab–MET complex was solved by molecular replacement with PHASER using previously solved MET Sema-PSI (PDB code 1SHY) and anti-HER3 Fab RG7116 (PDB code 4LEO) structures as search models. The structure was refined with PHENIX, and model adjustments were performed using COOT. His tags (at C-terminal of heavy chain and PSI), Fab interchain disulfide bond, heavy chain residues 133 to 139, Sema residues 303 to 309, 407, and glycan linked to N399 are disordered and not included in the structure. The Fab was numbered sequentially and Sema-PSI numbering starts at the N terminus of the signal peptide.
Epitope and paratope residues were assigned within a 4-Å contact distance cutoff using the CCP4 program CONTACT. The epitope area was calculated with the CCP4 program AREA. The buried surface area of binding residues was calculated with the program MOE (47
). Structural overlays of equivalent Cα atoms in the Sema domain (residues 40–515; PDB codes 1SHY, 4K3J, 2UZX, and 2UZY) were performed with COOT. Molecular graphics were generated with PyMol (PyMOL Molecular Graphics System, Version 1.4.1, Schrödinger, LLC) and MOE. The atomic coordinates and structure factors for the amivantamab anti-MET Fab–MET Sema-PSI complex were deposited in the RCSB PDB (accession code 6WVZ).
HCC827-HGF xenograft model
Female SCID Beige mice CB17.B6-Prkdcscid Lystbg/Crl (Charles River) bearing established subcutaneous HCC827-HGF tumors were randomized 13 days post inoculation (day 1). Individual tumor volumes ranged from 144 to 221 mm3; mean tumor volume ranged from 180 to 184 mm3. PBS and amivantamab (10 mg/kg) were dosed i.p. biweekly for 3 weeks. Crizotinib (30 mg/kg), erlotinib (25 mg/kg), crizotinib (30 mg/kg) and erlotinib (25 mg/kg), and vehicle controls (0.5% carboxymethyl cellulose in sterile water and 1% carboxymethyl cellulose in 0.1% Tween 80) were dosed daily p.o. for 3 weeks. Subcutaneous tumors were measured twice weekly as the mean tumor volume (mm3 ± standard error of the mean [SEM]). To calculate the percent tumor growth inhibition (%TGI) for group A versus group B, the tumor volumes were log transformed, where A = treated and B = control. The difference between these transformed values was taken at day 1 versus the designated day. Means were taken and converted by anti-log to numerical scale. Percentage TGIs were then calculated as (1 − A/B) × 100%. In vivo experiment was reviewed and approved by the Charles River Laboratories Institutional Animal Care and Use Committee and was done in accordance with the Guide for Care and Use of Laboratory Animals.
“Amivantamab”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT02609776 for “Study of Amivantamab, a Human Bispecific EGFR and cMet Antibody, in Participants With Advanced Non-Small Cell Lung Cancer (CHRYSALIS)” at ClinicalTrials.gov
To treat schizophrenia in adults and certain aspects of bipolar I disorder in adults
LYBALVI is a combination of olanzapine, an atypical antipsychotic, and samidorphan (as samidorphan L-malate), an opioid antagonist.
Olanzapine is 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5]benzodiazepine. The molecular formula of olanzapine is: C17H20N4S and the molecular weight is 312.44 g/mol. It is a yellow crystalline powder and has pKa values of 7.80 and 5.44. The chemical structure is:
Samidorphan L-malate is morphinan-3-carboxamide, 17-(cyclopropylmethyl)-4, 14-dihydroxy-6-oxo-, (2S)-2-hydroxybutanedioate. The molecular formula of samidorphan L-malate is C21H26N2O4 • C4H6O5 and the molecular weight is 504.54 g/mol. It is a white to off-white crystalline powder and has pKa values of 8.3 (amine) and 10.1 (phenol). The chemical structure is:
LYBALVI is intended for oral administration and is available as film-coated, bilayer tablets in the following strengths: 5 mg/10 mg, 10 mg/10 mg, 15 mg/10 mg, and 20 mg/10 mg of olanzapine and samidorphan (equivalent to 13.6 mg of samidorphan L-malate).
Inactive ingredients include colloidal silicon dioxide, crospovidone, lactose monohydrate, magnesium stearate, and microcrystalline cellulose. The film coating ingredients include hypromellose, titanium dioxide, triacetin, and color additives [iron oxide yellow (5 mg/10 mg); iron oxide yellow and iron oxide red (10 mg/10 mg); FD&C Blue No. 2/ indigo carmine aluminum lake (15 mg/10 mg); iron oxide red (20 mg/10 mg)].
to treat schizophrenia
alone for short-term (acute) or maintenance treatment of manic or mixed episodes that happen with bipolar I disorder
in combination with valproate or lithium to treat manic or mixed episodes that happen with bipolar I disorder
Olanzapine is an effective atypical antipsychotic that, like other antipsychotics, is associated with weight gain, metabolic dysfunction, and increased risk of type II diabetes.5,6 Samidorphan is a novel opioid antagonist structurally related to naltrexone, with a higher affinity for opioid receptors, more potent μ-opioid receptor antagonism, higher oral bioavailability, and a longer half-life, making it an attractive candidate for oral dosing.1,5,11 Although antipsychotic-induced weight gain is incompletely understood, it is thought that the opioid system plays a key role in feeding and metabolism, such that opioid antagonism may be expected to ameliorate these negative effects. Samidorphan has been shown in animal models and clinical trials to ameliorate olanzapine-induced weight gain and metabolic dysfunction.5,6
Samidorphan was first approved as a variety of fixed-dose combination tablets with olanzapine by the FDA on May 28, 2021, and is currently marketed under the trademark LYBALVI by Alkermes Inc.11
Samidorphan (INN, USAN) (developmental code names ALKS-33, RDC-0313), also known as 3-carboxamido-4-hydroxynaltrexone,[2] is an opioid antagonist that preferentially acts as an antagonist of the μ-opioid receptor (MOR). It is under development by Alkermes for the treatment of major depressive disorder and possibly other psychiatric conditions.[3]
However, it has attracted much more attention as part of the combination productALKS-5461 (buprenorphine/samidorphan), where samidorphan is combined with the mixed MOR weak partial agonist and κ-opioid receptor (KOR) antagonist buprenorphine, as an antidepressant. Buprenorphine has shown antidepressant effects in some human studies, thought to be because of its antagonist effects at the KOR, but has not been further developed for this application because of its MOR agonist effects and consequent abuse potential. By combining buprenorphine with samidorphan to block the MOR agonist effects, the combination acts more like a selective KOR antagonist, and produces only antidepressant effects, without typical MOR effects such as euphoria or substance dependence being evident.[6][7]
Samidorphan is also being studied in combination with olanzapine, as ALKS-3831 (olanzapine/samidorphan), for use in schizophrenia.[8] A Phase 3 study found that the addition of samidorphan to olanzapine significantly reduced weight gain compared to olanzapine alone.[9] The combination is now under review for approval by the US Food and Drug Administration.[10]
As such, samidorphan is primarily an antagonist, or extremely weak partial agonist of the MOR.[11][12] In accordance with its in vitro profile, samidorphan has been observed to produce some side effects that are potentially consistent with activation of the KOR such as somnolence, sedation, dizziness, and hallucinations in some patients in clinical trials at the doses tested.[13]
(A) Synthesis of 3-Carboxyamido-naltrexone 2[029] The triflate 11 of naltrexone was prepared according to the method of Wentland et al. (Bioorg. Med. Chem. Lett. 9, 183-187 (2000)), and the carboxamide 2 was prepared by the method described by Wentland et al. [(Bioorg. Med. Chem. Lett. ϋ, 623-626 (2001); and Bioorg. Med. Chem. Lett. 11, 1717-1721 (2001)] involving Pd-catalyzed carbonylation of the triflate 11 in the presence of ammonia and the Pd(O) ligand, DPPF ([l,l’-bis(diphenylρhosphino)ferrocene]) and DMSO.(B) Synthesis of 3-Carboxyamido-4-hydroxy-naltrexone derivative 3[030] Zinc dust (26 mg, 0.40 mmol) was added in portions to a solution of 2 (50 mg, 0.14 mmol) in HCl (37%, 0.2 mL) and AcOH (2 mL) at reflux. After heating at reflux for a further 15 min, the reaction was cooled by the addition of ice/water (10 mL) and basified (pH=9) with NH3/H2O, and the solution was extracted with EtOAc (3×10 mL). The organic extracts were washed with brine, dried, and concentrated. The residue was purified by column chromatography (SiO2, CH2Cl2, CH3OH : NH3/H2O = 15:1:0.01) to give compound 3 as a foam (25 mg, 50%). 1H NMR (CDC13) δl3.28(s, IH, 4-OH), 7.15(d, IH, J=8.1, H-2), 6.47(d, IH, J=8.4, H- 1), 6.10(br, IH, N-H), 4.35(br, IH, N-H), 4.04(dd,lH, J=I.8, 13.5, H-5), 3.11( d, IH, J=6), 2.99( d, IH, J=5.7), 2.94( s, IH), 2.86( d, IH, J= 6), 2.84-2.75(m, 2H), 2.65-2.61(m, 2H), 2.17-2.05(m, IH), 1.89-1.84(m, 2H), 0.85(m, IH), 0.56-0.50(m, 2H), 0.13-0.09(m, 2H). [α]D25= -98.4° (c=0.6, CH2Cl2). MS m/z (ESI) 371(MH+).
Opioid binding affinities were assessed for a series of cyclazocine analogues where the prototypic 8-OH substituent of cyclazocine was replaced by amino and substituted-amino groups. For μ and κ opioid receptors, secondary amine derivatives having the (2R,6R,11R)-configuration had the highest affinity. Most targets were efficiently synthesized from the triflate of cyclazocine or its enantiomers using Pd-catalyzed amination procedures.
In response to the unexpectedly high affinity for opioid receptors observed in a novel series of cyclazocine analogues where the prototypic 8-OH was replaced by a carboxamido group, we have prepared the corresponding 3-CONH2 analogues of morphine and naltrexone. High affinity (Ki=34 and 1.7 nM) for μ opioid receptors was seen, however, the new targets were 39- and 11-fold less potent than morphine and naltrexone, respectively.
Abstract
High-affinity binding to μ opioid receptors has been identified in a series of novel 3-carboxamido analogues of morphine and naltrexone.
References
^ Turncliff R, DiPetrillo L, Silverman B, Ehrich E (February 2015). “Single- and multiple-dose pharmacokinetics of samidorphan, a novel opioid antagonist, in healthy volunteers”. Clinical Therapeutics. 37 (2): 338–48. doi:10.1016/j.clinthera.2014.10.001. PMID25456560.
^ Wentland MP, Lu Q, Lou R, Bu Y, Knapp BI, Bidlack, JM (April 2005). “Synthesis and opioid receptor binding properties of a highly potent 4-hydroxy analogue of naltrexone”. Bioorganic & Medicinal Chemistry Letters. 15 (8): 2107–10. doi:10.1016/j.bmcl.2005.02.032. PMID15808478.
^ Hillemacher T, Heberlein A, Muschler MA, Bleich S, Frieling H (August 2011). “Opioid modulators for alcohol dependence”. Expert Opinion on Investigational Drugs. 20 (8): 1073–86. doi:10.1517/13543784.2011.592139. PMID21651459.
^ Clinical trial number NCT01366001 for “ALK33BUP-101: Safety and Pharmacodynamic Effects of ALKS 33-BUP Administered Alone and When Co-administered With Cocaine” at ClinicalTrials.gov
CAS Registry Number: 4759-48-2 CAS Name: 13-cis-Retinoic acid
Additional Names: 2-cis-vitamin A acid; neovitamin A acid
Manufacturers’ Codes: Ro-4-3780Trademarks: Accutane (Roche); Isotrex (Stiefel); Oratane (Douglas); Roaccutane (Roche) Molecular Formula: C20H28O2Molecular Weight: 300.44Percent Composition: C 79.95%, H 9.39%, O 10.65% Literature References: Naturally occurring metabolite of vitamin A, q.v.; inhibits sebum production. Prepn: C. D. Robeson et al.,J. Am. Chem. Soc.77, 4111 (1955). Stereoselective process: R. Lucci, EP111325; idem,US4556518 (1984, 1985 both to Hoffmann-La Roche). Toxicology and teratogenicity study: J. J. Kamm, J. Am. Acad. Dermatol.6, 652 (1982). Identification as endogenous metabolite of all-trans-retinoic acid: M. E. Cullum, M. H. Zile, J. Biol. Chem.260, 10590 (1985). HPLC determn in serum: G. Tang, R. M. Russell, J. Lipid Res.31, 175 (1990). Review of pharmacology and clinical efficacy in acne: A. R. Shalita et al.,Cutis42, Suppl. 6A, 1-19 (1988). Symposium on clinical experience: Dermatology195, Suppl. 1, 1-37 (1997). Properties: Reddish-orange plates from isopropyl alcohol, mp 174-175°. uv max: 354 nm (e 39800). LD50 (20 day) in mice, rats (mg/kg): 904, 901 i.p.; 3389, >4000 orally (Kamm).
Isotretinoin, also known as 13-cis-retinoic acid and sold under the brand name Accutane among others, is a medication primarily used to treat severe acne. It is also used to prevent certain skin cancers (squamous-cell carcinoma), and in the treatment of other cancers. It is used to treat harlequin-type ichthyosis, a usually lethal skin disease, and lamellar ichthyosis. It is a retinoid, meaning it is related to vitamin A, and is found in small quantities naturally in the body. Its isomer, tretinoin, is also an acne drug.
The most common adverse effects are a transient worsening of acne (lasting 1–4 months), dry lips (cheilitis), dry and fragile skin, and an increased susceptibility to sunburn. Uncommon and rare side effects include muscle aches and pains (myalgias), and headaches. Isotretinoin is known to cause birth defects due to in-utero exposure because of the molecule’s close resemblance to retinoic acid, a natural vitamin A derivative which controls normal embryonic development. It is also associated with psychiatric side effects, most commonly depression but also, more rarely, psychosis and unusual behaviours. Other rare side effects include hyperostosis, and premature epiphyseal closure, have been reported to be persistent.
In the United States, a special procedure is required to obtain the pharmaceutical. In most other countries, a consent form is required which explains these risks. In other countries, such as Israel, it is prescribed like any other medicine from a dermatologist (after proper blood tests).
Women taking isotretinoin must not get pregnant during and for one month after the discontinuation of isotretinoin therapy. Sexual abstinence or effective contraception is mandatory during this period. Barrier methods by themselves (e.g., condoms) are not considered adequate due to the unacceptable failure rates of approximately 3%. Women who become pregnant while taking isotretinoin therapy are generally counseled to have an abortion.
It was patented in 1969 and approved for medical use in 1982.[2] It sold well, but in 2009, Roche decided to discontinue manufacturing due to diminishing market share due to the availability of the many generic versions and the settling of multiple lawsuits over side effects. It continues to be manufactured as of 2019 by Absorica, Amnesteem, Claravis, Myorisan, Sotret, and Zenatane.[3]
Medical uses
Isotretinoin is used primarily for severe cystic acne and acne that has not responded to other treatments.[4][5][6][7] Many dermatologists also support its use for treatment of lesser degrees of acne that prove resistant to other treatments, or that produce physical or psychological scarring.[8] Isotretinoin is not indicated for treatment of prepubertal acne and is not recommended in children less than 12 years of age.[9]
Isotretinoin therapy has furthermore proven effective against genital warts in experimental use, but is rarely used for this indication as there are more effective treatments. Isotretinoin may represent an efficacious and safe alternative systemic form of therapy for recalcitrant condylomata acuminata (RCA) of the cervix. In most countries this therapy is currently unapproved and only used if other therapies failed.[11][12]
Prescribing restrictions
Isotretinoin is a teratogen; there is about a 20–35% risk for congenital defects in infants exposed to the drug in utero, and about 30–60% of children exposed to isotretinoin prenatally have been reported to show neurocognitive impairment.[13] Because of this, there are strict controls on prescribing isotretinoin to women who may become pregnant and women who become pregnant while taking isotretinoin are strongly advised to terminate their pregnancies.[13]
In most countries, isotretinoin can only be prescribed by dermatologists or specialist physicians; some countries also allow limited prescription by general practitioners and family doctors. In the United Kingdom[14] and Australia,[15][16] isotretinoin may be prescribed only by or under the supervision of a consultant dermatologist. Because severe cystic acne has the potential to cause permanent scarring over a short period, restrictions on its more immediate availability have proved contentious.[17] In New Zealand, isotretinoin can be prescribed by any doctor but subsidised only when prescribed by a vocationally-registered general practitioner, dermatologist or nurse practitioner.[18]
In the United States, since March 2006 the dispensing of isotretinoin is run through a website called iPLEDGE. The FDA required the companies marketing the drug in the US, which at the time that iPLEDGE was launched were Roche, Mylan, Barr, and Ranbaxy, to put this website in place as a risk evaluation and mitigation strategy. These companies formed a group called the Isotretinoin Products Manufacturing Group, and it hired Covance to run the website.[19][20] Prescribers, pharmacists, and all people to whom the drug is prescribed need to register on the site and log information into it. Women with child-bearing potential must commit to using two forms of effective contraception simultaneously for the duration of isotretinoin therapy and for a month immediately preceding and a month immediately following therapy. Additionally they must have two negative pregnancy tests 30 days apart and have negative pregnancy tests before each prescription is written.[21][22]
The compound 13-cis retinoic acid was first studied in the 1960s at Roche Laboratories in Switzerland by Werner Bollag as a treatment for skin cancer. Experiments completed in 1971 showed that the compound was likely to be ineffective for cancer and, surprisingly, that it could be useful to treat acne. However, they also showed that the compound was likely to cause birth defects, so in light of the events around thalidomide, Roche abandoned the product. In 1975, Gary Peck and Frank Yoder independently rediscovered the drug’s use as a treatment of cystic acne while studying it as a treatment for lamellar ichthyosis, and published that work. Roche resumed work on the drug. In clinical trials, subjects were carefully screened to avoid including women who were or might become pregnant. Roche’s New Drug Application for isotretinoin for the treatment of acne included data showing that the drug caused birth defects in rabbits. The FDA approved the application in 1982.
Scientists involved in the clinical trials published articles warning of birth defects at the same time the drug was launched in the US, but nonetheless isotretinoin was taken up quickly and widely, both among dermatologists and general practitioners. Cases of birth defects showed up in the first year, leading the FDA to begin publishing case reports and to Roche sending warning letters to doctors and placing warning stickers on drug bottles, and including stronger warnings on the label. Lawsuits against Roche started to be filed. In 1983 the FDA’s advisory committee was convened and recommended stronger measures, which the FDA took and were that time unprecedented: warning blood banks not to accept blood from people taking the drug, and adding a warning to the label advising women to start taking contraceptives a month before starting the drug. However use of the drug continued to grow, as did the number of babies born with birth defects. In 1985 the label was updated to include a boxed warning. In early 1988 the FDA called for another advisory committee, and FDA employees prepared an internal memo estimating that around 1,000 babies had been born with birth defects due to isotretinoin, that up to around 1,000 miscarriages had been caused, and that between 5,000 and 7,000 women had had abortions due to isotretinoin. The memo was leaked to the New York Times[77] a few days before the meeting, leading to a storm of media attention. In the committee meeting, dermatologists and Roche each argued to keep the drug on the market but to increase education efforts; pediatricians and the CDC argued to withdraw the drug from the market. The committee recommended to restrict physicians who could prescribe the drug and to require a second opinion before it could be prescribed. The FDA, believing it did not have authority under the law to restrict who had the right to prescribe the drug, kept the drug on the market but took further unprecedented measures: it required to Roche to make warnings yet more visible and graphic, provide doctors with informed consent forms to be used when prescribing the drug, and to conduct follow up studies to test whether the measures were reducing exposure of pregnant women to the drug. Roche implemented those measures, and offered to pay for contraception counseling and pregnancy testing for women prescribed the drug; the program was called the “Pregnancy Prevention Program”.
A CDC report published in 2000[78] showed problems with the Pregnancy Prevention Program and showed that the increase in prescriptions was from off-label use, and prompted Roche to revamp its program, renaming it the “Targeted Pregnancy Prevention Program” and adding label changes like requirements for two pregnancy tests, two kinds of contraception, and for doctors to provide pharmacists with prescriptions directly; providing additional educational materials, and providing free pregnancy tests. The FDA had another advisory meeting in late 2000 that again debated how to prevent pregnant women from being exposed to the drug; dermatologists testified about the remarkable efficacy of the drug, the psychological impact of acne, and demanded autonomy to prescribe the drug; others argued that the drug be withdrawn or much stricter measures be taken. In 2001 the FDA announced a new regulatory scheme called SMART (the System to Manage Accutane Related Teratogenicity) that required Roche to provide defined training materials to doctors, and for doctors to sign and return a letter to Roche acknowledging that they had reviewed the training materials, for Roche to then send stickers to doctors, which doctors would have to place on prescriptions they give people after they have confirmed a negative pregnancy test; prescriptions could only be written for 30 days and could not be renewed, thus requiring a new pregnancy test for each prescription.[citation needed]
In February 2002, Roche’s patents for isotretinoin expired, and there are now many other companies selling cheaper generic versions of the drug. On June 29, 2009, Roche Pharmaceuticals, the original creator and distributor of isotretinoin, officially discontinued both the manufacture and distribution of their Accutane brand in the United States due to what the company described as business reasons related to low market share (below 5%), coupled with the high cost of defending personal-injury lawsuits brought by some people who took the drug.[79] Generic isotretinoin will remain available in the United States through various manufacturers. Roche USA continues to defend Accutane and claims to have treated over 13 million people since its introduction in 1982. F. Hoffmann-La Roche Ltd. apparently will continue to manufacture and distribute Roaccutane outside of the United States.[80]
Among others, actor James Marshall sued Roche over allegedly Accutane-related disease that resulted in removal of his colon.[81] The jury, however, decided that James Marshall had a pre-existing bowel disease.[82]
Several trials over inflammatory bowel disease claims have been held in the United States thus far, with many of them resulting in multimillion-dollar judgments against the makers of isotretinoin.[83]
As of 2017 it was marketed as a topical combination drug with erythromycin under the brand names Isotrex Eritromicina, Isotrexin, and Munderm.[1]
Research
While excessive bone growth has been raised a possible side effect, a 2006 review found little evidence for this.[84]
syn
C. D. Robeson et al., J. Am. Chem. Soc. 77, 4111 (1955). Stereoselective process: R. Lucci, EP 111325; idem, US 4556518 (1984, 1985 both to Hoffmann-La Roche). doi:10.1021/jo00349a001.
syn
J Chem Soc 1968,(16),1982-83
The reaction of vinyl-beta-ionol (I) with triphenylphosphonium bromide (II) in ethanol gives the corresponding phosphonium salt (III), which is condensed through a Wittig reaction with cis-beta-formylcrotonic acid (IV) by means of sodium ethoxide in ethanol to afford a mixture of cis-2-cis-4-vitamin A acid (V) and the desired product. Finally, compound (V) is isomerized bv irradiation with diffuse light in ether in the presence of iodine.
syn
Tetrahedron 2000,56(37),7211
The formylation of the beta-ionone (I) with methyl formate and NaOMe gives the enol (II), which by reaction with methanol and H2SO4 yields the dimethylacetal (III). The reaction of (III) with methylenetriphenylphosphorane (IV) affords the methylene compound (V), which is treated with formic acid to provide the aldehyde (VI). The condensation of (VI) with isopropylidenemalonic acid dimethyl ester (VII) by means of NaOH gives the polyenic malonic acid (VIII) as a mixture of isomers that is separated by crystallization in ethyl ether to yield the desired all-trans-isomer (IX). Finally, this malonic acid is selectively monodecarboxylated by means of refluxing 2,6-dimethylpyridine to afford the target (E,E,E,Z)-isomer.
^ Strauss JS, Krowchuk DP, Leyden JJ, Lucky AW, Shalita AR, Siegfried EC, Thiboutot DM, Van Voorhees AS, Beutner KA, Sieck CK, Bhushan R (April 2007). “Guidelines of care for acne vulgaris management”. Journal of the American Academy of Dermatology. 56 (4): 651–63. doi:10.1016/j.jaad.2006.08.048. PMID17276540.
^ Sehgal VN, Srivastava G, Sardana K (June 2006). “Isotretinoin–unapproved indications/uses and dosage: a physician’s reference”. International Journal of Dermatology. 45 (6): 772–7. doi:10.1111/j.1365-4632.2006.02830.x. PMID16796650.
^ Specifically, doctors who are fellows of the Australasian College of Dermatologists (FACD); cf. Pharmaceutical Services Branch, Guide to poisons and therapeutic goods legislation for medical practitioners and dentists, Sydney: NSW Department of Health; 2006.[page needed]
^ DiGiovanna JJ (November 2001). “Isotretinoin effects on bone”. Journal of the American Academy of Dermatology. 45 (5): S176-82. doi:10.1067/mjd.2001.113721. PMID11606950.
^ Ellis CN, Madison KC, Pennes DR, Martel W, Voorhees JJ (1984). “Isotretinoin therapy is associated with early skeletal radiographic changes”. Journal of the American Academy of Dermatology. 10 (6): 1024–9. doi:10.1016/S0190-9622(84)80329-1. PMID6588057.
^ Scheinfeld N, Bangalore S (May 2006). “Facial edema induced by isotretinoin use: a case and a review of the side effects of isotretinoin”. Journal of Drugs in Dermatology. 5 (5): 467–8. PMID16703787.
^ Jump up to:abcd Borovaya A, Olisova O, Ruzicka T, Sárdy M (September 2013). “Does isotretinoin therapy of acne cure or cause depression?”. International Journal of Dermatology. 52 (9): 1040–52. doi:10.1111/ijd.12169. PMID23962262.
^ Jump up to:ab Rowe C, Spelman L, Oziemski M, Ryan A, Manoharan S, Wilson P, Daubney M, Scott J (May 2014). “Isotretinoin and mental health in adolescents: Australian consensus”. The Australasian Journal of Dermatology (Review). 55 (2): 162–7. doi:10.1111/ajd.12117. PMID24283385. S2CID29178483.
^ Goodman AB (May 1996). “Congenital anomalies in relatives of schizophrenic probands may indicate a retinoid pathology”. Schizophrenia Research. 19 (2–3): 163–70. doi:10.1016/0920-9964(96)88523-9. PMID8789914. S2CID12089905.
^ Lowenstein EB, Lowenstein EJ (2011). “Isotretinoin systemic therapy and the shadow cast upon dermatology’s downtrodden hero”. Clinics in Dermatology. 29 (6): 652–61. doi:10.1016/j.clindermatol.2011.08.026. PMID22014987.
^ Kremer I, Gaton DD, David M, Gaton E, Shapiro A (1994). “Toxic effects of systemic retinoids on meibomian glands”. Ophthalmic Research. 26 (2): 124–8. doi:10.1159/000267402. PMID8196934.
^ Griffin JN, Pinali D, Olds K, Lu N, Appleby L, Doan L, Lane MA (November 2010). “13-Cis-retinoic acid decreases hypothalamic cell number in vitro”. Neuroscience Research. 68 (3): 185–90. doi:10.1016/j.neures.2010.08.003. PMID20708044. S2CID207152111.
^ Sakai Y, Crandall JE, Brodsky J, McCaffery P (June 2004). “13-cis Retinoic acid (accutane) suppresses hippocampal cell survival in mice”. Annals of the New York Academy of Sciences. 1021 (1): 436–40. Bibcode:2004NYASA1021..436S. doi:10.1196/annals.1308.059. PMID15251924.
^ Jump up to:ab Peck GL, Olsen TG, Yoder FW, Strauss JS, Downing DT, Pandya M, Butkus D, Arnaud-Battandier J (February 1979). “Prolonged remissions of cystic and conglobate acne with 13-cis-retinoic acid”. The New England Journal of Medicine. 300 (7): 329–33. doi:10.1056/NEJM197902153000701. PMID153472.
^ Shalita A (2001). “The integral role of topical and oral retinoids in the early treatment of acne”. Journal of the European Academy of Dermatology and Venereology. 15: 43–9. doi:10.1046/j.0926-9959.2001.00012.x. PMID11843233.
^[unreliable medical source?]Farrell LN, Strauss JS, Stranieri AM (December 1980). “The treatment of severe cystic acne with 13-cis-retinoic acid. Evaluation of sebum production and the clinical response in a multiple-dose trial”. Journal of the American Academy of Dermatology. 3 (6): 602–11. doi:10.1016/S0190-9622(80)80074-0. PMID6451637.
^ Toyoda M, Nakamura M, Makino T, Kagoura M, Morohashi M (June 2002). “Sebaceous glands in acne patients express high levels of neutral endopeptidase”. Experimental Dermatology. 11 (3): 241–7. doi:10.1034/j.1600-0625.2002.110307.x. PMID12102663. S2CID23468315.
^ Wysowski DK, Swartz L (May 2005). “Relationship between headache and depression in users of isotretinoin”. Archives of Dermatology. 141 (5): 640–1. doi:10.1001/archderm.141.5.640. PMID15897395.
^ Ng CH, Schweitzer I (February 2003). “The association between depression and isotretinoin use in acne”. The Australian and New Zealand Journal of Psychiatry. 37 (1): 78–84. doi:10.1046/j.1440-1614.2003.01111.x. PMID12534661. S2CID8475675.
^ Halverstam CP, Zeichner J, Lebwohl M (2006). “Lack of significant skeletal changes after long-term, low-dose retinoid therapy: case report and review of the literature”. Journal of Cutaneous Medicine and Surgery. 10 (6): 291–9. doi:10.2310/7750.2006.00065. PMID17241599. S2CID36785828.
People diagnosed with trypanosome-caused disease should be treated with an anti-trypanosomal. Treatment is based on stage, 1 or 2, and parasite,T. b. rhodesiense or T. b. gambiense. In stage 1 disease, trypanosomes are present only in the peripheral circulation. In stage 2 disease, trypanosomes have crossed the blood-brain barrier and are present in the central nervous system.[6]
The following are considerable treatment options:[6]
Melarsoprol is a treatment used during the second stage of the disease. So far, it is the only treatment available for late-stage T. b. rhodesiense.[7]
Due to high toxicity, melarsoprol is reserved only for the most dangerous cases. Other agents associated with lower toxicity levels are used during stage 1 of the disease.[8] The approval of the nifurtimox-eflornithine combination therapy (NECT) in 2009 for the treatment of T. b. gambiense limited the use of melarsoprol to the treatment of second-stage T. b. rhodesiense.[9]
Failure rates of 27% in certain African countries have been reported.[10] This was caused by both drug resistance and additional mechanisms that have not yet been elucidated. Resistance is likely due to transport problems associated with the P2 transporter, an adenine-adenosine transporter. Resistance can occur with point mutations within this transporter.[11] Resistance has been present since the 1970s.[12]
Mechanism of action
Melarsoprol is a prodrug, which is metabolized to melarsen oxide (Mel Ox) as its active form. Mel Ox is an phenylarsonous acid derivative that irreversibly binds to sulfhydryl groups on pyruvate kinase, which disrupts energy production in the parasite. The inability to distinguish between host and parasite PK renders this drug highly toxic with many side effects.
Mel Ox also reacts with trypanothione (a spermidine-glutathione adduct that replaces glutathione in trypanosomes). It forms a melarsen oxide-trypanothione adduct (Mel T) that competitively inhibits trypanothione reductase, effectively killing the protist.[11]
Pharmacokinetics
The half-life of melarsoprol is less than one hour, but bioassays indicate a 35-hour half-life. This is commonly associated with pharmacologic agents that have active metabolites. One such metabolite, Mel Ox, reaches maximum plasma levels about 15 minutes after melarsoprol injection. Melarsoprol clearance is 21.5 ml/min/kg and the Mel Ox half-life is approximately 3.9 hours.[13]
Dosage
Two arsenic-containing stereoisomers exist in a 3:1 molar ratio. Since melarsoprol is insoluble in water, dosage occurs via a 3.6% propylene glycol intravenous injection.[11] To avoid the risk of injection site reactions, melarsoprol must be given slowly.[citation needed]
Melarsoprol used for the treatment of African trypanosomiasis with CNS involvement is given under a complicated dosing schedule. The dosing schedule for children and adults is 2–3.6 mg/kg/day intravenously for three days, then repeated every seven days for a total of three series.[6] To monitor for relapse, follow-up is recommended every six months for at least two years.[3]
Side effects
Although melarsoprol cures about 96% of people with late stage disease, its toxicity limits its use.[7] About 1-5% of people die during treatment.[3] As a toxic organic compound of arsenic, melarsoprol is a dangerous treatment that is typically only administered by injection under the supervision of a licensed physician. Notable side effects are similar to arsenic poisoning. Among clinicians, it is colloquially referred to as “arsenic in antifreeze”.[14] Severe and life-threatening adverse reactions are associated with melarsoprol. It is known to cause a range of side effects including convulsions, fever, loss of consciousness, rashes, bloody stools, nausea and vomiting. In approximately 5-10% of cases, it causes encephalopathy. Of those, about 50% die due to encephalopathy-related adverse reactions.[6] Additional potentially serious side effects of melarsoprol include damage to the heart, presence of albumin in the urine that could be associated with kidney damage, and an increase in blood pressure.[3]
Cautions
Numerous warnings must be examined before melarsoprol treatment can be initiated. Prior to initiation, the following must be noted: glucose-6-phosphate dehydrogenase deficiency, kidney or liver disease, cardiac problems (high blood pressure, irregular beating of the heart or arrhythmias, any damage to the heart muscles and potential signs of heart failure), preexisting nervous system disorders, and any signs of leprosy.
Routine laboratory testing is needed before and after melarsoprol initiation. Laboratory parameters for both therapeutic effects and toxic effects need to be evaluated.
Blood analysis is used to detect the presence of trypanosomes. An evaluation of the cerebrospinal fluid via a lumbar puncture is also used to determine an individual’s white blood count and level of protein. These are diagnostic criteria such that the presence of trypanosomes, an elevated white blood count greater than five per microliter, or a protein content greater than 40 mg are considered abnormal and initiation should be considered. Continuous cerebrospinal fluid evaluation should be repeated every six months for at least three years in individuals that have undergone melarsoprol treatment.
To assess potential concerns related to toxicity, the following should be completed: a complete blood count, an assessment of electrolyte levels, liver and kidney function tests, and a urinalysis to detect the appearance, concentration and content of the urine.
Melarsoprol should be given using glass syringes (if they can be reliably sterilised). The propylene glycol it contains is capable of dissolving plastic.[15]
Pregnancy and breastfeeding
Currently, melarsoprol is not recommended for use in pregnant women. The World Health Organization suggests that treatment be deferred until immediately after delivery since the effects of the medication on the developing fetus have not yet been established.[3]
Lactation guidelines associated with melarsoprol have not yet been established.
Society and culture
Melarsoprol is produced by Sanofi-Aventis and under an agreement with the WHO, they donate melarsoprol to countries where the disease is common.[medical citation needed]
Melarsoprol was used to treat a patient with African trypanosomiasis on season 1 episode 7 “Fidelity” of the medical drama House MD.[16]
PAPER
Journal of Organometallic Chemistry (2006), 691(5), 1081-1084.
(2-Phenyl-[1,3,2]dithiarsolan-4-yl)-methanol derivatives were tested on K562 and U937 human leukemia cell lines. Their systemic toxicity was estimated by the corresponding LD50 on mice. The cytotoxic activity of each derivative was significantly better than that of arsenic trioxide and the therapeutic index (T.I. = LD50/IC50) was improved.
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Jump up to:abc Brunton L (2011). “Goodman & Gillman’s The Pharmacological Basis of Therapeutics”. McGraw Hill Medical: 1427–28.
^ Brun, Reto; Schumacher, Reto; Schmid, Cecile; Kunz, Christina; Burri, Christian (November 2001). “The phenomenon of treatment failures in Human African Trypanosomiasis”. Tropical Medicine and International Health. 6 (11): 906–914. doi:10.1046/j.1365-3156.2001.00775.x. PMID11703845. S2CID21542129.
^ Keiser J.; Ericsson O; Burri C (2000). “Investigations of the metabolites of the trypanocidal drug melarsoprol”. Clinical Pharmacology. 67 (5): 478–88. doi:10.1067/mcp.2000.105990. PMID10824626. S2CID24326873.
Ezutromid, also known as BMN-195 and SMTC-1100, is a first orally bioavailable utrophin’s translation modulator. Duchenne muscular dystrophy (DMD) is a lethal, progressive muscle wasting disease caused by a loss of sarcolemmal bound dystrophin, which results in the death of the muscle fibers leading to the gradual depletion of skeletal muscle.
Ezutromid is an orally administered small molecule utrophin modulator currently involved in a Phase 2 clinical trial produced by Summit Therapeutics for the treatment of Duchenne muscular dystrophy (DMD).[1][2] DMD is a fatal x-linked recessive disease affecting approximately 1 in 5000 males and is a designated orphan disease by the FDA and European Medicines Agency.[3] Approximately 1/3 of the children obtain DMD as a result of spontaneous mutation in the dystrophin gene and have no family history of the disease.[3]Dystrophin is a vital component of mature muscle function, and therefore DMD patients have multifarious forms of defunct or deficient dystrophin proteins that all manifest symptomatically as muscle necrosis and eventually organ failure.[3][4] Ezutromid is theorized to maintain utrophin, a protein functionally and structurally similar to dystrophin that precedes and is replaced by dystrophin during development.[3][5] Utrophin and dystrophin are reciprocally expressed, and are found in different locations in a mature muscle cell.[4][6] However, in dystrophin-deficient patients, utrophin was found to be upregulated and is theorized to replace dystrophin in order to maintain muscle fibers.[7] Ezutromid is projected to have the potential to treat all patients suffering with DMD as it maintains the production of utrophin to counteract the lack of dystrophin to retard muscle degeneration.[7][8] Both the FDA and European Medicines Agency has given ezutromid an orphan drug designation.[5][9] The FDA Office of Orphan Products and Development offers an Orphan Drug Designation program (ODD) that allows drugs aimed to treat diseases that affect less than 200,000 people in the U.S. monetary incentives such as a period of market exclusivity, tax incentives, and expedited approval processes.[5][10]
The Phase 2 clinical trial was ended in 2018 and the medication discontinued after it failed to show any benefit in slowing the disease.[11]
Clinical trials
The first Phase 1b trial (NCT02056808) began on November 2013 and involved 12 patients aged 5–11 years old.[12] The patients were divided into three groups given escalating oral doses testing the safety and tolerability after each increase over the course of 10 days.[12]
Another completed Phase 1b trial (NCT02383511) began February 2015 and involved 12 patients aged 5–13 years old.[13] The goal was to determine the safety, tolerability, and pharmacokinetic parameters by measuring plasma concentration and major metabolite levels over 28 days for three sequence groups.[13] Each sequence involved placebo, 1250 mg, and 2500 mg BID (twice a day) doses given for one week each.[4][13]
A PhaseOut DMD, Phase 2, Proof of Concept (NCT02858362) clinical trial is underway that tests the clinical safety and efficacy of an oral suspension of ezutromid.[2] The 48-week open-label trial is enrolling 40 boys, ages 5–10, living in the U.K. or U.S.[2] MRI leg muscle change will be measured as well as ezutromid plasma concentration levels, with a secondary goal of obtaining quantifiable images of utrophin membrane stained biopsies at baseline and either 24 or 48 weeks.[2]
Commercial aspects
As of 2016, ataluren was the only approved drug in the EU to treat a specific subpopulation of patients with nmDMD, or DMD caused by a nonsense mutation.[14] However, nonsense mutations only account for approximately 15% of all patients with DMD.[15] Therefore, Summit Therapeutics projects to file for regulatory approval in the US and EU by 2019 and to reach market in 2020.[8] They expect to profit just over £24,046 in 2020 and £942,656 in 2025, which amounts to ~10% CGR for the first 7 years on the basis of treating all DMD patients in the US, EU, Iceland, Norway, Switzerland and Russia.[8]
Furthermore, Summit Therapeutics has entered an agreement with Sarepta Theraputics as of October 2016 regarding the commercialization of ezutromid.[16] The agreement consists of a collaboration between Sarepta and Summit to share the research and developing costs for the development of novel therapies to treat DMD patients.[16]
4-(ethylthio)Phenol S2: To a 250 mL round bottle, 4-mercaptophenol S1 (12.6 g, 100 mmol), K2CO3 (15.3 g, 110 mmol), acetone (100 mL) were added, then, iodoethane (15.6 g, 8.0 mL, 130 mmol) was added slowly at 0 oC. The system was stirred at room temperature overnight. After filtration, distillation of solvent, and flash chromatography, S2 (10.780 g) was obtained with 70% yield.
4-(ethylthio)-2-Nitrophenol S3: To a 250 mL round bottle, 4-(ethylthio)Phenol S2 (3.084 g, 20 mmol), 300-400 mesh silica gel (2 g), distilled water (2 g), and CH3CN (60 mL) was added. The system was then cooled by an ice water bath. Subsequently, citric acid (3.842 g, 20 mmol), NaNO2 (2.760 g, 40 mmol) were separately added slowly in portionwise. The system was reacted at room temperature overnight. After filtration and distillation of solvent, EA (50 mL) and water (50 mL) was added, after separation, the aqueous phase was extraction with EA (30 mL) twice. The combined organic phase was dried with MgSO4. Following by filtration and chromatography, S3 (3.590 g) was obtained with 90% yield.
4-(ethylthio)-2-Nitrophenol S4: To a 100 mL round bottle, S3 (2.46 g, 12.3 mmol), reductive iron powder (2.07 g, 36.9 mmol), and EtOH (50 mL) was added. Then, HCl (aq.) (0.15 M) (12 mL, 1.85 mmol) was added slowly. The system was refluxed overnight. After filtration, distillation of solvent, and flash chromatography, S4 (1.040 g) was obtained with 50% yield.
5-(ethylthio)-2-(naphthalen-2-yl)Benzo[d]oxazole S6 (Ezutromid-S): S4 (324 mg1.91 mmol), 2-naphthoyl chloride S5 (545.7 mg, 2.87 mmol), dry 1,4-dioxane (5 mL) was added into a sealing tube. Then, the system was vacuumed and filled with nitrogen for three times. Subsequently, the reaction was run at 160 oC for 10 hours. After distillation of solvent and flash chromatography, S6 (361.7 mg) was obtained with 62% yield. 1H NMR (500 MHz, Chloroform-d) δ 8.74 (s, 1H), 8.28 (dd, J = 8.5, 1.7 Hz, 1H), 7.96 (t, J = 7.5 Hz, 2H), 7.92 – 7.84 (m, 1H), 7.81 (d, J = 1.8 Hz, 1H), 7.57 (pd, J = 6.8, 3.4 Hz, 2H), 7.51 (d, J = 8.3 Hz, 1H), 7.39 (dd, J = 8.4, 1.8 Hz, 1H), 2.99 (q, J = 7.3 Hz, 2H), 1.33 (t, J = 7.3 Hz, 3H).13C NMR (126 MHz, Chloroform-d) δ 163.79, 149.70, 142.92, 134.76, 132.89, 132.38, 128.92, 128.78, 128.20, 127.87, 127.85, 126.91, 124.11, 123.84, 121.38, 110.72, 29.17, 14.40
5- (ethylthio)-2-(naphthalen-2-yl)Benzo[d]oxazole (30.5 mg, 0.1 mmol), UO2(OAc)2 . 2H2O (0.8 mg, 0.002 mol), H2O (10 equiv., 36 μL), o-xylene (8.3 equiv., 0.2 mL), CH3CN (1 mL) were stirred under oxygen atmosphere (1 atm, balloon) at room temperature until the total consumption of sulfide and sulfoxide under the irradiation of three 2 w blue LEDs in a paralleled reactor. 5a (27.3 mg, 81%) was obtained through column chromatography (PE/EA = 20/1-5/1) as a white solid, Rf = 0.6 (PE/EA = 2/1);
It has been discovered that the compound of formula I (5-(ethylsulfonyl)-2-(naphthalen-2-yl)benzo[d]oxazole) has excellent properties for the treatment of Duchenne muscular dystrophy (see, e.g., international patent application publication no. WO 2007/091106).
The compound of formula I (R = 5-ethylsulfonyl; R9 = 2-naphthalen-2-yl) may be synthesised according to the following procedure, as disclosed in WO 2007/091106 (page 51):
Experimental
S nthesis of 5- eth lsulfon -2- na hthalen-2- l‘benzo d oxazole
Procedure: A vessel was equipped with a retreat blade stirrer and downward pumping turbine, a five necked flange lid, seal and clamp, stirrer gland and overhead stirrer, thermometer pocket, Dean- Stark trap, dropping funnel and condenser. The water to the condenser was then switched on. The sodium hydroxide and 0.80 L of water were then mixed (whilst cooling in an ice bath until all the sodium hydroxide has dissolved – caution exothermic). The resulting solution was then transferred to a scrubber appropriately attached to the vessel.
The 2-amino-4-(ethylsulfonyl)phenol and 2.00 L of xylenes (mixed) were then transferred to the vessel, and the reagents and solvent were stirred at 100 rpm. Then, the 2-naphtholyl chloride was dissolved in 2.00 L of xylenes (mixed) and transferred into the vessel. The stirring rate was increased to 150 rpm.
The temperature of the solution was gradually increased to 100°C over a period of not less than 30 mins, and then maintained at that level for 10 mins. (Caution: HCl gas is evolved during this process through the gas scrubber). The stirrer speed was then increased to 315 rpm and the temperature gradually increased over a period of 30 minutes until reflux (155°C) at which level it was maintained for 90 mins. (Caution: HCl gas is evolved during this process through the gas scrubber). The methanesulfonic acid was then added drop-wise over a period of 30 mins and relux was maintained until no further water was being collected in the Dean-Stark apparatus (approx 15 mins). The heat was then removed and the pipe adapter from the Dean- Stark apparatus disconnected. The resulting solution was allowed to cool to 900C, and then filtered using Whatman 1 filter paper. The resulting solution was then left at ambient temperature for 18h, after which time the product crystallised, and the product was separated by filtration using Whatman 1 filter paper. The product was then washed with Ix 1.0 L of tert-butyl methyl ether (TBME)
The product was then dried in a vacuum oven at 65°C at a pressure of 1 Ombar until constant weight was achieved (less than 0.5 g difference between consecutive measurements of mass which must be at least 1 h apart). The product was obtained as a sandy-beige powder in a yield of 80%.
1. Procedure 100 mg of the compound of formula I was dissolved in the minimum amount of good solvent and then the anti-solvent was added to induce crystallisation. The supernatant liquor was then removed, and the resulting solid was dried under vacuum for 12 his.
PAPER
Journal of medicinal chemistry (2011), 54(9), 3241-50
A series of novel 2-arylbenzoxazoles that upregulate the production of utrophin in murine H2K cells, as assessed using a luciferase reporter linked assay, have been identified. This compound class appears to hold considerable promise as a potential treatment for Duchenne muscular dystrophy. Following the delineation of structure–activity relationships in the series, a number of potent upregulators were identified, and preliminary ADME evaluation is described. These studies have resulted in the identification of 1, a compound that has been progressed to clinical trials.
PAPER
Angewandte Chemie, International Edition (2019), 58(38), 13499-13506
Angewandte Chemie, International Edition (2020), 59(3), 1346-1353.
Journal of medicinal chemistry (2020), 63(5), 2547-2556.
Abstract
5-(Ethylsulfonyl)-2-(naphthalen-2-yl)benzo[d]oxazole (ezutromid, 1) is a first-in-class utrophin modulator that has been evaluated in a phase 2 clinical study for the treatment of Duchenne muscular dystrophy (DMD). Ezutromid was found to undergo hepatic oxidation of its 2-naphthyl substituent to produce two regioisomeric 1,2-dihydronaphthalene-1,2-diols, DHD1 and DHD3, as the major metabolites after oral administration in humans and rodents. In many patients, plasma levels of the DHD metabolites were found to exceed those of ezutromid. Herein, we describe the structural elucidation of the main metabolites of ezutromid, the regio- and relative stereochemical assignments of DHD1 and DHD3, their de novo chemical synthesis, and their production in systems in vitro. We further elucidate the likely metabolic pathway and CYP isoforms responsible for DHD1 and DHD3 production and characterize their physicochemical, ADME, and pharmacological properties and their preliminary toxicological profiles.
Following on from ezutromid, the first-in-class benzoxazole utrophin modulator that progressed to Phase 2 clinical trials for the treatment of Duchenne muscular dystrophy, a new chemotype was designed to optimise its physicochemical and ADME profile. Herein we report the synthesis of SMT022357, a second generation utrophin modulator preclinical candidate, and an asymmetric synthesis of its constituent enantiomers. The pharmacological properties of both enantiomers were evaluated in vitro and in vivo. No significant difference in the activity or efficacy was observed between the two enantiomers; activity was found to be comparable to the racemic mixture.
^ Jump up to:abc Werther CA (2016). Ezutromid Has the Potential to Treat All Duchenne Patients; Initiating Coverage With a Buy. H.C. Wainwright & Co. pp. 1–29.
^ Jump up to:abc Clinical trial number NCT02383511 for “Modified Diet Trial: A Study of SMT C1100 in Paediatric Patients With DMD Who Follow a Balanced Diet ” at ClinicalTrials.gov
Bemiparin is an ultra-low molecular weight heparin (ultra-LMWH) used to prevent thromboembolism following surgery and extracorporeal clotting during dialysis.
Rovi and Archimedes (a wholly owned subsidiary of ProStrakan), have developed and launched bemiparin, a Factor Xa inhibitor for the injectable treatment and prevention of thrombosis.
low or very low molecular weight heparins (eg bemiparin sodium) with a high anti-factor Xa activity for the treatment of deep vein thrombosis.
Bemiparin is an antithrombotic and belongs to the group of drugs known as the low molecular weight heparins (LMWH). Like semuloparin, bemiparin is classified as an ultra-LMH because of its low mean molecular mass of 3600 daltons, which is a unique property of this class 1. These heparins have lower anti-thrombin activity than the traditional low molecular weight heparins and act mainly on factor-Xa, reducing the risk of bleeding due to selectivity for this specific clotting factor. Interestingly, current research is underway for the potential benefit of bemiparin in the treatment of tumors and diabetic foot ulcers 12,1.
Laboratorios Farmaceuticos Rovi has developed and launched Enoxaparina Rovi, a biosimilar version of enoxaparin sodium, an injectable low-molecular-weight fraction of heparin, for the prophylaxis of venous thromboembolism.
claiming a method for analyzing glycosaminoglycans, heparins and their derivatives in a compound comprising a monosaccharide residues present in heparin (eg bemiparin sodium) chains by identification and relative quantification of its characteristic signals by1H NMR one-dimensional nuclear magnetic resonance and/or 1H-13C HSQC two-dimensional nuclear magnetic resonance, using dimethylmalonic acid as internal reference
PATENT
CN-110092848
https://patents.google.com/patent/CN110092848A/enEmbodiment 1Experimental raw used and instrument are as follows in embodiment 1:Refined heparin sodium (ZH160712 quality of lot meets CP2015), benzethonium chloride, purified water, 40% (W/V) trimethoxy Base methanolic ammonium hydroxide, methylene chloride, methanol, 10% (W/V) sodium acetate methanol solution, 30% hydrogen peroxide, medicinal second Alcohol, sodium chloride, glass reaction pot (5000ml) three-necked flask 500ml, digital display heat-collecting magnetic stirring device, beaker, freeze dryer (on Hai Dongfulong) etc..A kind of preparation method of Bemiparin sodium of the present invention, the following steps are included:1. at salt1.1 weigh, dissolution, react1.1.1 the refined heparin sodium for weighing 10g is poured into tank, and the purified water of 100ml is added into reactor tank, is stirred to molten Solution is complete.1.1.2 25g benzethonium chloride is added in beaker, 125ml purified water stirring and dissolving is added.1.1.3 benzethonium chloride solution is added slowly with stirring in the heparin sodium aqua in reactor tank, time for adding 4.5h controls 35 DEG C of feed liquid temperature, continues stirring 2 hours, stops stirring and stands 2 hours, then as far as possible by supernatant liquid Removing.1.2 washings, centrifugation, drying:1.2.1 300ml purified water is added into residue precipitating suspended matter to wash in three times, then starts to wash for the first time, 20 DEG C of feed liquid temperature of control is stirred 1 hour, is stopped stirring and is stood 2 hours, repeats the above operation twice.1.2.2 supernatant liquid is removed, filters and be washed with water under stirring, record slurry amount, collect sediment.1.2.3 final gained sediment is uniformly divided in stainless steel disc, is transferred in heated-air circulation oven, adjust temperature 40 DEG C of degree, dry 6h crushes solid with Universalpulverizer after then 60 DEG C of dry range estimations are not glued to solid, smashed solid Body continues to be transferred in heated-air circulation oven, until loss on drying≤2.0%.Rewinding obtains heparin-benzyl rope ammonium salt about 32g, does Dry weightless 1.5%.2. degradation2.1 weighingBy above-mentioned 30g heparin-benzyl rope ammonium salt in 500ml three-necked flask, the methylene chloride of 150ml is added into reactor tank It is added in three-necked flask.2.2 dissolutions: three-necked flask is put into digital display heat-collecting magnetic stirring device, is heated to 33 DEG C and is stirred to having dissolved Entirely.2.3 degradations: being added 40% (W/V) trimethoxy methanolic ammonium hydroxide of 20.4ml in Xiang Shangshu solution, puts down Respectively 4 additions, it is for 24 hours that interval time is added every time.It after the 4th is added, then reacts for 24 hours, amounts to reaction 96h, during reaction Maintain 34 DEG C of temperature.2.4 terminate reaction: above-mentioned reaction solution being cooled to 20 DEG C, 180ml10% (W/V) sodium acetate methanol is added thereto Solution stirs 30min, filters to obtain its precipitating.2.5 washings: washing above-mentioned sediment with 300ml methanol solution, dry bemiparin crude product about 9g.3. purification3.1 will be above-mentioned dry that 9g bemiparin crude product pours into tank, and the purified water of 90ml, stirring are added into reactor tank It is complete to dissolution.3.2 adjust material liquid pH 9.5 with 20% sodium hydroxide solution.0.54ml hydrogen peroxide is added to be stirred to react at 20 DEG C 7.5 hours, through 0.22 μm of micro porous filtration.3.3 1.8g sodium chloride is added into feed liquid, then uses 4mol/L hydrochloric acid flavouring liquid pH to 6.5, is added into feed liquid 450ml medicinal alcohol stops stirring after stirring 30 minutes, places 4 hours.3.4 take supernatant away, and 90ml purified water is added, and stirring adjusts PH6.5 to dissolving completely, through 0.22 μm of micro porous filtration, Sabot freeze-drying.After 3.5 freeze-drying 36h, collection material weighing 7g.Three, the primary quality measure statistics of gained bemiparin
Serial number
Project
Control standard
Testing result
1
Weight average molecular weight
3000~4200
3650
2
Molecular weight is greater than 6000 constituent content
< 15%
12.9%
3
Constituent content of the molecular weight less than 2000
< 35%
36.7%
4
Molecular weight is between 2000~6000 constituent contents
50%~75%
50.4%
5
Anti-Xa activity
80~120IU/mg
116IU/mg
6
Anti- IIa activity
5~20IU/mg
14.6IU/mg
7
The anti-anti- IIa of Xa/
≥7
7.95
Embodiment 2Experimental raw used and instrument are as follows in embodiment 1:Refined heparin sodium (ZH180912 quality of lot meets CP2015), benzethonium chloride, purified water, 40% (W/V) trimethoxy Base methanolic ammonium hydroxide, methylene chloride, methanol, 10% (W/V) sodium acetate methanol solution, 30% hydrogen peroxide, medicinal second Alcohol, sodium chloride, glass reaction pot (10000ml, 30000L), three-necked flask 500ml, digital display heat-collecting magnetic stirring device, beaker, Freeze dryer (Shanghai Dong Fulong) etc..A kind of preparation method of Bemiparin sodium of the present invention, the following steps are included: 1. one-tenth salt1.1 weigh, dissolution, react1.1.1 the refined heparin sodium for weighing 500g is poured into tank, the purified water of 5000ml is added into reactor tank, stirring is extremely Dissolution is complete.1.1.2 1250g benzethonium chloride is added in beaker, 6300ml purified water stirring and dissolving is added.1.1.3 benzethonium chloride solution is added slowly with stirring in the heparin sodium aqua in reactor tank, time for adding 5h controls 35 DEG C of feed liquid temperature, continues stirring 2 hours, stops stirring and stands 2 hours, then as far as possible by supernatant liquid It removes.1.2 washings, centrifugation, drying:1.2.1 5000ml purified water is added into residue precipitating suspended matter to wash in three times, then starts to wash for the first time, 30 DEG C of feed liquid temperature of control is stirred 1 hour, is stopped stirring and is stood 2 hours, repeats the above operation twice.1.2.2 supernatant liquid is removed, filters and be washed with water under stirring, record slurry amount, collect sediment.1.2.3 final gained sediment is uniformly divided in stainless steel disc, is transferred in heated-air circulation oven, adjust temperature 45 DEG C of degree, dry 6h crushes solid with Universalpulverizer after then 70 DEG C of dry range estimations are not glued to solid, smashed solid Body continues to be transferred in heated-air circulation oven, until loss on drying≤2.0%.Rewinding obtains heparin-benzyl rope ammonium salt about 1505g, Loss on drying 1.0%.2. degradation2.1 weighingBy above-mentioned 1500g heparin-benzyl rope ammonium salt in 30L glass reaction kettle, the methylene chloride of 7500ml is added thereto.2.2 dissolutions: leading to hot water for its interlayer, is heated to 33~36 DEG C and stirs complete to dissolving.2.3 degradations: being added 40% (W/V) trimethoxy methanolic ammonium hydroxide of 1020ml in Xiang Shangshu solution, puts down Respectively 4 additions, it is for 24 hours that interval time is added every time.It after the 4th is added, then reacts for 24 hours, amounts to reaction 96h, during reaction Maintain 35 DEG C of temperature.2.4 terminate reaction: above-mentioned reaction solution being cooled to 20 DEG C, 9000ml10% (W/V) sodium acetate first is added thereto Alcoholic solution stirs 30min, filters to obtain its precipitating.2.5 washings: washing above-mentioned sediment with 15000ml methanol solution, dry bemiparin crude product about 400g.3. purification3.1 will be above-mentioned dry that 400g bemiparin crude product pours into tank, and the purified water of 4000ml is added into reactor tank, Stirring is complete to dissolving.3.2 adjust material liquid pH 9.5 with 20% sodium hydroxide solution.24ml hydrogen peroxide is added, and at 30 DEG C to be stirred to react 7 small When, through 0.22 μm of micro porous filtration.3.3 8g sodium chloride is added into feed liquid, then uses 4mol/L hydrochloric acid flavouring liquid pH to 6.5, is added into feed liquid 20000ml medicinal alcohol stops stirring after stirring 30 minutes, places 4 hours.3.4 take supernatant away, and 4000ml purified water is added, and stirring adjusts PH6.5, through 0.22 μm of micropore mistake to dissolving completely Filter, sabot freeze-drying.After 3.5 freeze-drying 36h, collection material weighing 350g.Three, the primary quality measure statistics of gained bemiparin
Heparin belongs to the glycosaminoglycan family and is a polysaccharide of animal origin, which is extracted from the intestine or lungs of mammals (cow, lamb, pig) and is used in human therapies for the prevention and treatment of thromboembolic diseases . It is well known that the use of heparin is accompanied by very annoying bleeding effects and its daily administration, three subcutaneous or intravenous injections, constitutes a very considerable inconvenience.
During the course of the last few years, different chemical methods have been used to depolymerize heparin, such as:
– treatment with sodium nitrite in an acid medium,
– alkaline treatment of asters,
– use of free radicals generated in the presence of hydrogen peroxide,
– treatment of a quaternary ammonium salt of heparin in a non-aqueous medium with a strong base according to a beta elimination mechanism.
These methods make it possible to obtain, with variable yields, mixtures of heparin fragments in which the average molecular weight and anticoagulant activity vary according to the procedure and operating conditions. Low molecular weight heparins (LMWH) described in the state of the art or commercialized are obtained according to different depolymerization procedures. Their average molecular weights (Mw) are in the range of 3,600 and 7,500 Daltons.
It is now recognized that the antithrombotic activity of LMWH is mainly due to its ability to activate antithrombin III, a plasma protein and potent inhibitor of activated factor X and thrombin. In this way, it is possible to measure the antithrombotic activity of heparin by means of specific tests to determine the inhibition of these factors.
Research carried out by different authors shows that heparin fragments or oligosaccharides, with short chains of average molecular weight <4,800 Daltons, have a selective action on activated factor X and not on thrombin, in determinations using methods of the Pharmacopoeia. .
It has been found that if very low molecular weight fragments are required that have strong anti-factor Xa activity, it is preferable to use a selective depolymerization technique in non-aqueous medium, as described in US patent 9,981,955, which respects the antithrombin III binding site.
The document EP 1070503 A1 describes the controlled depolymerization of heparin using a process in a non-aqueous medium that makes it possible to obtain a family of LMWH that are obtained enriched in low molecular weight oligosaccharides that have a high anti-factor Xa activity and a low anti-factor lia activity, and which can be represented by the general formula:
in which:
n can vary between 1 and 12,
Ri = H or S0 3 Na,
R 2 = SOsNao COCH 3 ,
Said very low molecular weight heparin is obtained by selective depolymerization of heparin in a non-aqueous medium according to a beta elimination procedure.
No interaction studies have been conducted. Drugs that are expected to increase the risk of bleeding in combination with bemiparin include other anticoagulants, aspirin and other NSAIDs, antiplatelet drugs, and corticosteroids.[2]
Chemistry
Like semuloparin, bemiparin is classified as an ultra-LMWH because of its low molecular mass of 3600 g/mol on average.[3] (Enoxaparin has 4500 g/mol.) These heparins have lower anti-thrombin activity than classical LMWHs and act mainly on factor Xa, reducing the risk of bleeding.[4]
References
^ Chapman TM, Goa KL (2003). “Bemiparin: a review of its use in the prevention of venous thromboembolism and treatment of deep vein thrombosis”. Drugs. 63 (21): 2357–77. doi:10.2165/00003495-200363210-00009. PMID14524738.
^ Jump up to:abcAustria-Codex (in German). Vienna: Österreichischer Apothekerverlag. 2018. Ivor 2500 IE Anti-Xa/0,2 ml Injektionslösung in Fertigspritzen.
^ Planès A (September 2003). “Review of bemiparin sodium–a new second-generation low molecular weight heparin and its applications in venous thromboembolism”. Expert Opinion on Pharmacotherapy. 4 (9): 1551–61. doi:10.1517/14656566.4.9.1551. PMID12943485. S2CID13566575.
^ Jeske WP, Hoppensteadt D, Gray A, Walenga JM, Cunanan J, Myers L, Fareed J, Bayol A, Rigal H, Viskov C (October 2011). “A common standard is inappropriate for determining the potency of ultra low molecular weight heparins such as semuloparin and bemiparin”. Thrombosis Research. 128 (4): 361–7. doi:10.1016/j.thromres.2011.03.001. PMID21458847.
Chapman TM, Goa KL: Bemiparin: a review of its use in the prevention of venous thromboembolism and treatment of deep vein thrombosis. Drugs. 2003;63(21):2357-77. [Article]
Planes A: Review of bemiparin sodium–a new second-generation low molecular weight heparin and its applications in venous thromboembolism. Expert Opin Pharmacother. 2003 Sep;4(9):1551-61. [Article]
Jeske WP, Hoppensteadt D, Gray A, Walenga JM, Cunanan J, Myers L, Fareed J, Bayol A, Rigal H, Viskov C: A common standard is inappropriate for determining the potency of ultra low molecular weight heparins such as semuloparin and bemiparin. Thromb Res. 2011 Oct;128(4):361-7. doi: 10.1016/j.thromres.2011.03.001. Epub 2011 Apr 2. [Article]
Hoffman M, Monroe DM: Coagulation 2006: a modern view of hemostasis. Hematol Oncol Clin North Am. 2007 Feb;21(1):1-11. doi: 10.1016/j.hoc.2006.11.004. [Article]
Antonijoan RM, Rico S, Martinez-Gonzalez J, Borrell M, Valcarcel D, Fontcuberta J, Barbanoj MJ: Comparative pharmacodynamic time-course of bemiparin and enoxaparin in healthy volunteers. Int J Clin Pharmacol Ther. 2009 Dec;47(12):726-32. [Article]
Publication numberPriority datePublication dateAssigneeTitleUS4981955A *1988-06-281991-01-01Lopez Lorenzo LDepolymerization method of heparinEP0293539B1 *1987-01-051994-06-08Laboratorios Farmaceuticos Rovi, S.A.Process for the depolymerization of heparin for obtaining heparin with a low molecular weight and having an antithrombotic activityCN1379781A *1999-10-222002-11-13阿文蒂斯药物股份有限公司Novel oligosaccharides, preparation method and pharmaceutical composition containing sameCN102399306A *2010-09-092012-04-04上海喜恩医药科技发展有限公司Preparation method of heparin-derived polysaccharide mixtureCN105693886A *2016-04-192016-06-22常州市蓝勖化工有限公司Preparation method of heparin sodiumCN106467577A *2015-08-212017-03-01苏州融析生物科技有限公司A kind of pulmonis Bovis seu Bubali Enoxaparin Sodium and preparation method and applicationCN106977627A *2017-05-162017-07-25苏州二叶制药有限公司A kind of Enoxaparin production method of sodiumCN109575156A *2018-11-052019-04-05上海宝维医药技术有限公司A kind of purification process of low molecular weight heparinFamily To Family Citations
PropofolCAS Registry Number: 2078-54-8 CAS Name: 2,6-Bis(1-methylethyl)phenolAdditional Names: 2,6-diisopropylphenol; disoprofol Manufacturers’ Codes: ICI-35868 Trademarks: Ansiven (Abbott); Diprivan (AstraZeneca); Disoprivan (AstraZeneca); Rapinovet (Schering-Plough Vet.)Molecular Formula: C12H18OMolecular Weight: 178.27Percent Composition: C 80.85%, H 10.18%, O 8.97% Literature References: Prepn: A. J. Kolka et al.,J. Org. Chem.21, 712 (1956); 22, 642 (1957); G. G. Ecke, A. J. Kolka, US2831898 (1958 to Ethyl Corp.); T. J. Kealy, D. D. Coffman, J. Org. Chem.26, 987 (1961); B. E. Firth, T. J. Rosen, US4447657 (1984 to Universal Oil Products). Chromatographic study: J. K. Carlton, W. C. Bradbury, J. Am. Chem. Soc.78, 1069 (1956). Animal studies: J. B. Glen, Br. J. Anaesth.52, 731 (1980).Pharmacokinetics: H. K. Adam et al.,ibid. 743; idem,ibid.55, 97 (1983). Determn in blood: eidem,J. Chromatogr.223, 232 (1981). Comparative studies vs other injectable anesthetics: B. Kay, D. K. Stephenson, Anaesthesia35, 1182 (1980); D. V. Rutter et al.,ibid. 1188. Use in i.v. anesthesia: E. Major et al.,ibid.37, 541 (1982). Cardiovascular effects: D. Al-Khudhairi et al.,ibid. 1007. Pharmacology of emulsion formulation: J. B. Glen, S. C. Hunter, Br. J. Anaesth.56, 617 (1984). Series of articles on pharmacology and clinical experience: Postgrad. Med. J.61, Suppl. 3, 1-169 (1985). Properties: bp30 136°. bp17 126°. mp 19°. nD20 1.5134. nD25 1.5111. d20 0.955.Melting point: mp 19°Boiling point: bp30 136°; bp17 126°Index of refraction:nD20 1.5134; nD25 1.5111Density: d20 0.955Therap-Cat: Anesthetic (intravenous).Therap-Cat-Vet: Intravenous anesthetic (dogs and cats).Keywords: Anesthetic (Intravenous).SYN
Prepn: A. J. Kolka et al., J. Org. Chem. 21, 712 (1956); 22, 642 (1957); G. G. Ecke, A. J. Kolka, US 2831898 (1958 to Ethyl Corp.); T. J. Kealy, D. D. Coffman, J. Org. Chem. 26, 987 (1961); B. E. Firth, T. J. Rosen, US 4447657 (1984 to Universal Oil Products).SYN
A commercially viable manufacturing process for propofol (1) is described. The process avoids acid–base neutralization events during isolation of intermediate, 2,6-di-isopropylbenzoic acid (3) and crude propofol, and thus simplifies the synthesis on industrial scale to a considerable extent. Syntheses of five impurities/related substances (USP and EP) are also described.
Propofol is used during surgeries for sedation and an injectable grade with purity > 99.90% is desired by the medical community. An embodiment of the present invention provides an economically feasible, industrial process for the manufacture of high purity injectable grade Propofol. An embodiment of the present invention relates to a process and novel strategy for purification of 2,6-diisopropylphenol (Propofol) and similar products.
[0003] Propofol is a sterile injectable drug that appears in the USP, EP and IP Monographs. Drug product is manufactured by using high purity drug substance 2,6-di-isopropylphenol commonly known as Propofol.
[0004] Propofol is used to put patients to sleep and keep them asleep during general anesthesia for surgery or other medical procedures. It is used in adults as well as children 2 months and older. Propofol is frequently used as a sedative, and has a rapid onset of action and a short recovery period. Propofol slows the activity of brain and nervous system. Propofol is also used to sedate a patient who is under critical care and needs a mechanical ventilator (breathing machine). Propofol is a hypnotic alkylphenol derivative. When formulated for intravenous induction of sedation and hypnosis during anaesthesia, Propofol facilitates inhibitory neurotransmission mediated by gamma- Aminobutyric acid (GABA). Propofol is associated with minimal respiratory depression and has a short half-life with a duration of action of 2 to 10 minutes.
[0005] Propofol is commonly used parenteral anesthetic agent in the United States, extensively used for minor and outpatient surgical procedures because of its rapid onset and reversal of action, and in intensive care units (ICUs) for maintaining coma. Propofol has been associated with rare instances of idiosyncratic acute liver injury; in addition, prolonged high dose Propofol therapy can cause the “Propofol infusion syndrome” which is marked by brady arrhythmias, metabolic acidosis, rhabdomyolysis, hyperlipidemia and an enlarged or fatty liver.
[0006] Friedel-Craft’s alkylation of phenol using propylene gas in the presence of Lewis acid (LA) catalysts is a commonly used method for the synthesis of Propofol and is well documented in a number of publications and patents [Ecke, G. G., Kolka, A. J. US 2,831,898 A, 1958. Firth, B. E., Rosen, T. J. US 4,447,657, 1984. Akio, T., Yoshiaki, I., Hidekichi, H., Kiyoji, K., Takashi, K., Masanobu, M. EP 0169359A1, 1986. Ecke, G. G., Kolka, A. J. US 3,271,314, 1966. Napolitano, J. P. US 3,367,981 A, 1968. Goddard L. E. US 3,766,276, 1973. Firth, B. E. US 4,275,248, 1981, etc.]
[0007] A number of patents and published literature describe the manufacture of Propofol. US. Pat. No. 4,275,248; W0200034218; EP169359; US. Pat. No. 3,367,981; US. Pat. No.
3,271,314; US. Pat. No. 3,766,276; US. Pat. No. 2,831,898; US.Pat.No.2,207,753; GB1318100; U.S. Pat. No. 4,391,998; US. Pat. No. 4,774, 368; US. Pat. No. 5,589,598; US. Pat. No. 6,362,234; etc. EP 0511947, discloses purification of Propofol that is obtained by alkylation of phenol and purified by crystallization at -10 to -20°C (melting point of Propofol is 18°C). This patent also describes purification using non-polar solvents such as Petroleum ether or Hexane, where solvent residue is removed by distillation or evaporation and finally Propofol is obtained using fractional distillation under high vacuum.
[0008] Continuous separation of a mixture of Propofol with phenolic impurities and methanol is described in an U.S. Pat. No. 5,264,085. U.S. Pat. No. 5,705,039 describes the purification of impure 2,6-diisopropylphenol first using continuous distillation and then distilling pure Propofol under high vacuum.
[0009] Patent CN103360219A describes purification wherein 2,6-diisopropyl phenol is reacted with benzoyl chloride to generate ‘benzoic acid-2, 6-diisopropyl benzene ester’, which is then purified to yield Propofol. The patent discloses that an adsorbent is added at the rectifying stage, so that impurities with similar chemical structures and boiling points are effectively removed; the content of a single impurity in the product is not higher than 0.01%; the total impurity is not higher than 0.05%.
[0010] CN105601477A describes purification of Propofol wherein crude Propofol is purified with three-stage distillation method; the crude Propofol enters feeding tank protected by nitrogen and is charged into first-stage film distillation system through pump; then the product is fed to second-stage molecular distillation system and low boiling point impurities are removed; finally, the processed product is charged into third-stage molecular distiller through a pump, high-boiling-point impurities are separated, and the colourless or yellowish high-purity Propofol is obtained.
[0011] In another prior art disclosure, after completion of the reaction, the final product is isolated and purified by high-vacuum distillation. Alkylation of phenol using propylene gas at high pressure and high temperature is reported. Several impurities like 2,4-diisopropyl and 2,4,6-triisopropyl phenol are the major side products along with the corresponding Isopropyl ether. All these impurities need to be controlled at a limit of NMT 0.05% or less in the final API for it to be pharmaceutically acceptable. In another prior art disclosure, isopropanol was used as the propylating agent instead of direct propylene gas. In this method propylene is generated in situ using IPA and strong acid like sulfuric acid and catalysts like Aluminoslicate [See Baltalksne, A. E.; Zitsmanis, A. H. SU 443019, 1974. Jain, K. P., Edaki, D. U., Minhas H. S., Minhas G. S. WO/2011/ 161687 Al, 2011. Wu, M. US 4,391,998, 1983]
[0012] Another method is to use of protected phenol, where 4-chloro or 4-carboxylic acid substituted phenol is used as starting material along with Isopropanol in sulfuric acid, followed by removal of the 4-substituent to give Propofol [Baltalksne, A. E.; Zitsmanis, A. H. SU 443019, 1974. Jain, K. P., Edaki, D. U., Minhas H. S., Minhas G. S. WO/2011/ 161687 Al, 2011. Wu, M. US 4,391,998, 1983. Tsutsumi, S.; Yoshizawa, T.; Koyama, K. Nippon Kagaku Zasshi 1956, 77, 737-738. Paiocchi, M. US 5,589,598, 1996. Nieminen, K., Essen, P. US 5,175,376, 1992. Keller, S., Schlegel, J. WO/2012/152665 Al, 2012.] The final purification is carried out by high- vacuum distillation to get highly pure Propofol. Since the para position is blocked, related impurities such as 2,4-isopropyl and 2,4,6-triisopropyl derivatives are avoided. In this approach, intermediate is purified before converting to crude Propofol using either de-chlorination by hydrogenation or de-carboxylation before vacuum distillation for final purification.
[0013] It is reported in the literature that 4-hydroxybenzoic acid is used as starting material for alkylation with isopropyl alcohol in sulfuric acid. In that method 2,6-diisopropyl-4-hydroxy benzoic acid gets formed, which is extracted in toluene either in presence of an acid or the impurities are extracted in toluene under alkaline condition. The decarboxylation is carried out using solvents like monoethylene glycol or ethoxyethanol at high temperature. At the end of decarboxylation, crude Propofol is isolated by extracting into toluene. The advantage is Propofol does not form sodium salt under the conditions, but all other acidic impurities form sodium salt and thus do not get extracted in toluene. The toluene containing Crude Propofol is distilled to recover toluene and then vacuum distilled to obtain pure Propofol. [Chen, T; Chen, X.; Bois-Choussy, M.; Zhu, J. J. Am. Chem. Soc. 2006, 128, 87-89. Lau, S.; Keay, B. Can. J. Chem. 2001, 79, 1541-1545]
[0014] In summary, strategies disclosed in prior art for the production of 2,6-diisopropylphenol (Propofol) predominantly involve synthesis starting from phenol or by using protected 4-position of phenol like, 4-hydroxybenzoic acid, 4-chlorophenol (references: Baltalksne, A. E.; Zitsmanis, A. H. SU 443019, 1974. Jain, K. P., Edaki, D. U., Minhas H. S., Minhas G. S. WO/2011/ 161687 Al, 2011. Wu, M. US 4,391,998, 1983. Tsutsumi, S.; Yoshizawa, T.; Koyama, K. Nippon Kagaku Zasshi 1956, 77, 737-738. Paiocchi, M. US 5,589,598, 1996. Nieminen, K., Essen, P. US 5,175,376, 1992. Keller, S., Schlegel, J. WO/2012/152665 Al, 2012). Processes described in the literature generally propose purification of crude 2,6-diisopropylphenol by ‘high vacuum distillation or molecular distillation’.
[0015] The phenols are susceptible to oxidation, formation of polymeric and color impurities. There are processes where repeated vacuum distillation has been carried out to obtain desired purity of product. Sometimes, to reduce the oxidized and colored impurities, reduction of impurities by catalytic hydrogenation is also used.
[0016] Propofol that does not meet Pharmaceutical grade may be manufactured by several processes generally known to persons of skill in the art and described in prior art, but purification of Propofol to consistently achieve high purity required for the injectable drug substance using an economical and industrial process remains a challenge.
Example 1:
[0033] Commercially available concentrated sulfuric acid (30 Kg) was diluted with water (2.26 Kg) at low temperature (0-15°C). Methyl 4-hydroxybenzoate (5 Kg 32.79 mol.) was added to this diluted sulfuric acid at 5 to 10 °C with stirring. After complete addition, isopropyl alcohol (5.9 Kg 98.16 mol.) was gradually added to the reaction content, controlling the temperature at 0-15 °C. The reaction mixture was then heated at 60-70°C and continued to complete di-isopropylation and ester hydrolysis to yield methyl-4-hydroxybenzoate. The conversion was checked on TLC or by HPLC for the complete conversion of methyl-4 hydroxybenzoate to 3, 5 -Diisopropyl 4-hydroxybenzoic acid.
[0034] The reaction contents were cooled at room temperature and carefully charged into a stirred, precooled mixture of water (50 L) and Toluene (40 L) at (0 to 5°C). The mixture was stirred and maintained below 15°C for about 30 to 60 minutes.
[0035] The content was then heated at 25 to 30°C, stirred for 30 min., allowed to settle into two layers. The water layer was extracted again with toluene and discarded. The toluene layers, containing the product 3, 5-Diisopropyl 4-hydroxybenzoic acid, were combined and extracted with about 25 L of 10 % NaOH. The aqueous layer containing the sodium salt of 3, 5 -Di-isopropyl 4-hydroxybenzoic acid was acidified with concentrated HC1 (about 9 Kg) to precipitate 3, 5-Diisopropyl 4-hydroxybenzoic acid, filtered, and washed with water (about 50 L) to yield 3, 5 -diisopropyl 4-hydroxybenzoic acid (about 45-60 %)
[0036] To the mixture of 3, 5-diisopropyl 4-hydroxybenzoic acid (3 Kg, 13.5 mol.) in ethylene glycol (5.0 Kg, 80.55 mol.) was added sodium hydroxide (1.25 Kg, 31.25 mol.) for decarboxylation. The reaction mixture was heated at 145 ± 5°C till completion of
decarboxylation by monitoring using TLC or HPLC (or solubility in bicarbonate of precipitated product). After complete decarboxylation, the reaction mixture was cooled at 40 to 45 °C, under nitrogen environment and diluted with water (about 15 L) and allowed to settle. The oily product layer was separated and washed with water (6L) to isolate crude Propofol (i.e., 2,6-diisopropyl phenol) and stored under nitrogen. The isolated volatile Crude Propofol (along with carry over ppm ethylene glycol and NaOH) was then subjected to steam distillation purification process as described below.
[0037] The Crude Propofol is purified by using one of the steam distillation processes as described below.
[0038] The Crude Propofol layer is added to purified water in a reactor (preferably glass lined reactor), and slowly heated to boiling to co-distil Pure Propofol along with water under normal atmospheric pressure and the high volatile initial fraction is isolated first. The biphasic layers of main distillate, are separated and the liquid layer of Propofol is treated with dehydrating agent to absorb dissolved moisture in Pure Propofol under nitrogen or argon. The transparent Pure Propofol liquid layer is then filtered through ultrafme Micron filter (0.45 and 0.2 micron) under nitrogen (or argon) pressure and isolated in controlled environment to obtain pharmaceutical injectable grade Pure Propofol of more than 99.90% purity.
[0039] The Crude Propofol liquid layer is charged into a reactor with steam distillation arrangement, like steam purging dip tube, column, heat exchanger and receivers. Pure steam is purged in the reactor at controlled pressure to co-distil Pure Propofol with water. The layers are allowed settle and water layer is kept aside for recirculation. The transparent Pure Propofol transparent liquid layer is then filtered through ultrafme Micron filter (0.45 and 0.2 micron) under nitrogen (or argon) pressure and isolated in controlled environment to obtain pharmaceutical injectable grade Pure Propofol of more than 99.90% purity.
[0040] The Crude Propofol layer is added to purified water in a reactor (preferably glass lined or Hastelloy reactor) and slowly heated at boiling to co-distil Pure Propofol along with water under mild vacuum. The biphasic layers are separated and the liquid layer of Propofol is treated with dehydrating agent to absorb dissolved moisture in Pure Propofol under nitrogen (or argon). The transparent Pure Propofol liquid layer is then filtered through ultrafme Micron filter (0.45 and 0.2 micron) under nitrogen (or argon) pressure and isolated in controlled environment to obtain pharmaceutical injectable grade Pure Propofol of more than 99.90% purity.
[0041] The Crude Propofol layer is added to reactor containing purified water and 0.1 to 1% antioxidant and 0.1 to 0.5% sodium hydroxide and slowly heated to boiling to co-distil Pure Propofol along with water. The biphasic layers are separated and the liquid layer of Propofol is treated or passed through column packed with dehydrating agent to absorb dissolved moisture in Pure Propofol. The transparent Pure Propofol liquid layer is then filtered through ultrafme Micron filter (0.45 and 0.2 micron) under nitrogen (or argon) pressure and isolated in controlled environment to obtain pharmaceutical injectable grade Pure Propofol of more than 99.90% purity.
[0042] The crude Propofol liquid layer is treated with preferably neutral or basic activated carbon (about 2-5%) and filtered under nitrogen. The filtered liquid is collected, under nitrogen, in distillation reactor containing purified water is slowly heated to boiling to co-distil Pure Propofol along with water under normal pressure or mild vacuum. The co-distilled biphasic layers are separated and the liquid layer of Propofol, is treated under nitrogen, with or passed through column packed with dehydrating agent to absorb dissolved moisture trapped in Pure Propofol. The transparent Pure Propofol liquid layer is then filtered through ultrafme Micron filter (0.45 and 0.2 micron) under nitrogen (or argon) pressure and isolated in controlled environment to obtain pharmaceutical injectable grade Pure Propofol of more than 99.90% purity.
Example No. 2:
[0043] Friedel-Crafts reaction was performed as described in Example 1. Decarboxylation was performed by using KOH instead of NaOH by following the same procedure as described in Example 1.
Example No. 3:
[0044] Decarboxylation was performed as per operations described in Example 1. After complete decarboxylation, the reaction mixture was cooled at 40 to 45°C, under nitrogen environment and diluted with water (about 15 L) The biphasic mixture subjected to steam distillation by any of the purification methods described in Example 1.
Example No. 4:
[0045] Friedel-Crafts reaction was performed as described in Example 1. The reaction contents were cooled at room temperature and carefully charged at 0 to 5°C into a sodium hydroxide solution to basic pH at stirred. The aqueous alkaline solution was extracted twice with toluene or hexane. The aqueous layer was acidified with HC1 to precipitate 3, 5-diisopropyl-4-hydroxybenzoic acid. The wet product was washed with water, dried and decarboxylated using sodium hydroxide in ethylene glycol as solvent at 145±5°C. The reaction contents were cooled to room temperature, diluted with water and acidified and then Crude Propofol was extracted twice in toluene. The toluene layer was washed with water, bicarbonate and with water then distilled to obtain crude oily layer of Propofol (>99% pure). This Crude Propofol was then purified by using purification steam distillation by any of the purification methods described in Example 1.
Example 5:
[0046] Continuous steam distillation of crude Propofol by purging pure steam. Continuous steam distillation of Crude Propofol was carried out using a feed pump for feeding liquid Crude Propofol (prepared by one of the processes described in this application or other literature) to the steam distillation system connected to a pure steam generator. Steam at 1-10 kg pressure was purged in the steam distillation system at controlled rate and the co-distilled Pure Propofol with water was cooled using heat exchanger and continuous separator. The residue was discharged from bottom valve at defined time intervals. The water layer was recycled to steam generator and Pure Propofol was dehydrated, filtered and collected in controlled environment as described in Example 1.
Propofol, marketed as Diprivan, among other names, is a short-acting medication that results in a decreased level of consciousness and a lack of memory for events.[4] Its uses include the starting and maintenance of general anesthesia, sedation for mechanically ventilated adults, and procedural sedation.[4] It is also used for status epilepticus if other medications have not worked.[4] It is given by injection into a vein, and the maximum effect takes about two minutes to occur and typically lasts five to ten minutes.[4] Propofol is also used for medical assistance in dying in Canada.[5]
To induce general anesthesia, propofol is the drug used almost always, having largely replaced sodium thiopental.[14][6] It can also be administered as part of an anesthesia maintenance technique called total intravenous anesthesia, using either manually programmed infusion pumps or computer-controlled infusion pumps in a process called target controlled infusion (TCI). Propofol is also used to sedate individuals who are receiving mechanical ventilation but not undergoing surgery, such as patients in the intensive care unit.[15][16] In critically ill patients, propofol is superior to lorazepam both in effectiveness and overall cost.[17] Propofol is relatively inexpensive compared to medications of similar use due to shorter ICU stay length.[17] One of the reasons propofol is thought to be more effective (although it has a longer half-life than lorazepam) is because studies have found that benzodiazepines like midazolam and lorazepam tend to accumulate in critically ill patients, prolonging sedation.[17] Propofol has also been suggested as a sleep aid in critically ill adults in the ICU, however, the effectiveness of this medicine at replicating the mental and physical aspects of sleep for people in the ICU are not clear.[16]
Propofol is often used instead of sodium thiopental for starting anesthesia because recovery from propofol is more rapid and “clear”.[citation needed]
Propofol can be run through a peripheral IV or central line. Propofol is frequently paired with fentanyl (for pain relief) in intubated and sedated people.[18] Both are compatible in IV form.[18]
Procedural sedation
Propofol is also used for procedural sedation. Its use in these settings results in a faster recovery compared to midazolam.[19] It can also be combined with opioids or benzodiazepines.[20][21][22] Because of its rapid induction and recovery time, propofol is also widely used for sedation of infants and children undergoing MRI.[23] It is also often used in combination with ketamine with minimal side effects.[24]
COVID-19
In March 2021, the U.S. Food and Drug Administration (FDA) issued an emergency use authorization (EUA) for Propofol‐Lipuro 1% to maintain sedation via continuous infusion in people greater than age sixteen with suspected or confirmed COVID‑19 who require mechanical ventilation in an intensive care unit ICU setting.[25][26][27][28] In the circumstances of this public health emergency, it would not be feasible to require healthcare providers to seek to limit Fresenius Propoven 2% Emulsion or Propofol-Lipuro 1% only to be used for patients with suspected or confirmed COVID‑19; therefore, this authorization does not limit use to such patients.[28]
Other uses
Executions
The US state of Missouri added propofol to its execution protocol in April 2012. However, Governor Jay Nixon halted the first execution by the administration of a lethal dose of propofol in October 2013 following threats from the European Union to limit the drug’s export if it were used for that purpose.[29][30] The United Kingdom had already banned the export of medicines or veterinary medicines containing propofol to the United States.[31]
Recreational use
Recreational use of the drug via self-administration has been reported[32][33] but is relatively rare due to its potency and the level of monitoring required for safe use.[citation needed] Critically, a steep dose-response curve makes recreational use of propofol very dangerous, and deaths from self-administration continue to be reported.[34][35] The short-term effects sought via recreational use include mild euphoria, hallucinations, and disinhibition.[36][37]
Recreational use of the drug has been described among medical staff, such as anesthetists who have access to the drug.[38][39] It is reportedly more common among anesthetists on rotations with short rest periods, as usage generally produces a well-rested feeling.[40] Long-term use has been reported to result in addiction.[38][41]
One of propofol’s most common side effects is pain on injection, especially in smaller veins. This pain arises from activation of the pain receptor, TRPA1,[48] found on sensory nerves and can be mitigated by pretreatment with lidocaine.[49] Less pain is experienced when infused at a slower rate in a large vein (antecubital fossa). Patients show considerable variability in their response to propofol, at times showing profound sedation with small doses.
Additional side effects include low blood pressure related to vasodilation, transient apnea following induction doses, and cerebrovascular effects. Propofol has more pronounced hemodynamic effects relative to many intravenous anesthetic agents.[50] Reports of blood pressure drops of 30% or more are thought to be at least partially due to inhibition of sympathetic nerve activity.[51] This effect is related to the dose and rate of propofol administration. It may also be potentiated by opioid analgesics.[52] Propofol can also cause decreased systemic vascular resistance, myocardial blood flow, and oxygen consumption, possibly through direct vasodilation.[53] There are also reports that it may cause green discolouration of the urine.[54]
Although propofol is heavily used in the adult ICU setting, the side effects associated with propofol seem to be of greater concern in children. In the 1990s, multiple reported deaths of children in ICUs associated with propofol sedation prompted the FDA to issue a warning.[55]
As a respiratory depressant, propofol frequently produces apnea. The persistence of apnea can depend on factors such as premedication, dose administered, and rate of administration, and may sometimes persist for longer than 60 seconds.[56] Possibly as the result of depression of the central inspiratory drive, propofol may produce significant decreases in respiratory rate, minute volume, tidal volume, mean inspiratory flow rate, and functional residual capacity.[50]
Diminishing cerebral blood flow, cerebral metabolic oxygen consumption, and intracranial pressure are also characteristics of propofol administration.[57] In addition, propofol may decrease intraocular pressure by as much as 50% in patients with normal intraocular pressure.[58]
Propofol is also reported to induce priapism in some individuals,[60][61] and has been observed to suppress REM sleep stage and to worsen the poor sleep quality in some patients.[62]
As with any other general anesthetic agent, propofol should be administered only where appropriately trained staff and facilities for monitoring are available, as well as proper airway management, a supply of supplemental oxygen, artificial ventilation, and cardiovascular resuscitation.[63]
Because of its lipid base, some hospital facilities require the IV tubing (of continuous propofol infusions) to be changed after 12 hours. This is a preventive measure against microbial growth and infection.[64]
A rare, but serious, side effect is propofol infusion syndrome. This potentially lethal metabolic derangement has been reported in critically ill patients after a prolonged infusion of high-dose propofol, sometimes in combination with catecholamines and/or corticosteroids.[65]
Propofol has been proposed to have several mechanisms of action,[67][68][69] both through potentiation of GABAA receptor activity and therefore acting as a GABAA receptor positive allosteric modulator, thereby slowing the channel-closing time. At high doses, propofol may be able to activate GABAA receptors in the absence of GABA, behaving as a GABAA receptor agonist as well.[70][71][72] Propofol analogs have been shown to also act as sodium channel blockers.[73][74] Some research has also suggested that the endocannabinoid system may contribute significantly to propofol’s anesthetic action and to its unique properties.[75]EEG research upon those undergoing general anesthesia with propofol finds that it causes a prominent reduction in the brain’s information integration capacity.[76]
Pharmacokinetics
A 20 ml ampoule of 1% propofol emulsion, as sold in Australia by Sandoz
Propofol is highly protein-bound in vivo and is metabolised by conjugation in the liver.[77] The half-life of elimination of propofol has been estimated to be between 2 and 24 hours. However, its duration of clinical effect is much shorter, because propofol is rapidly distributed into peripheral tissues. When used for IV sedation, a single dose of propofol typically wears off within minutes. Propofol is versatile; the drug can be given for short or prolonged sedation, as well as for general anesthesia. Its use is not associated with nausea as is often seen with opioid medications. These characteristics of rapid onset and recovery along with its amnestic effects[78] have led to its widespread use for sedation and anesthesia.
History
John B. Glen, a veterinarian and researcher at Imperial Chemical Industries (ICI) spent 13 years developing propofol, an effort which led to the awarding to him of the prestigious 2018 Lasker Award for clinical research. Propofol was originally developed as ICI 35868. It was chosen for development after extensive evaluation and structure–activity relationship studies of the anesthetic potencies and pharmacokinetic profiles of a series of ortho-alkylated phenols.[79]
First identified as a drug candidate in 1973, clinical trials followed in 1977, using a form solubilised in cremophor EL.[80] However, due to anaphylactic reactions to cremophor, this formulation was withdrawn from the market and subsequently reformulated as an emulsion of a soya oil/propofol mixture in water. The emulsified formulation was relaunched in 1986 by ICI (now AstraZeneca) under the brand name Diprivan. The currently available preparation is 1% propofol, 10% soybean oil, and 1.2% purified egg phospholipid as an emulsifier, with 2.25% glycerol as a tonicity-adjusting agent, and sodium hydroxide to adjust the pH. Diprivan contains EDTA, a common chelation agent, that also acts alone (bacteriostatically against some bacteria) and synergistically with some other antimicrobial agents. Newer generic formulations contain sodium metabisulfite or benzyl alcohol as antimicrobial agents. Propofol emulsion is a highly opaque white fluid due to the scattering of light from the tiny (about 150-nm) oil droplets it contains: Tyndall Effect.
Developments
A water-soluble prodrug form, fospropofol, has been developed and tested with positive results. Fospropofol is rapidly broken down by the enzyme alkaline phosphatase to form propofol. Marketed as Lusedra, this formulation may not produce the pain at injection site that often occurs with the conventional form of the drug. The U.S. Food and Drug Administration (FDA) approved the product in 2008.[81] However fospropofol is a Schedule IV controlled substance with the DEA ACSCN of 2138 in the United States unlike propofol.[82]
By incorporation of an azobenzene unit, a photoswitchable version of propofol (AP2) was developed in 2012, that allows for optical control of GABAA receptors with light.[83] In 2013, a propofol binding site on mammalian GABAA receptors has been identified by photolabeling using a diazirine derivative.[84] Additionally, it was shown that the hyaluronan polymer present in the synovia can be protected from free-radicaldepolymerization by propofol.[85]
^ Ruffle JK (November 2014). “Molecular neurobiology of addiction: what’s all the (Δ)FosB about?”. Am J Drug Alcohol Abuse. 40 (6): 428–437. doi:10.3109/00952990.2014.933840. PMID25083822. S2CID19157711. Propofol is a general anesthetic, however its abuse for recreational purpose has been documented (120). Using control drugs implicated in both ΔFosB induction and addiction (ethanol and nicotine), similar ΔFosB expression was apparent when propofol was given to rats. Moreover, this cascade was shown to act via the dopamine D1 receptor in the NAc, suggesting that propofol has abuse potential (119)
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Jump up to:ab Isert, Peter R.; Lee, Doris; Naidoo, Daya; Carasso, Melanie L.; Kennedy, Ross A. (June 1996). “Compatibility of propofol, fentanyl, and vecuronium mixtures designed for potential use in anesthesia and patient transport”. Journal of Clinical Anesthesia. 8 (4): 329–336. doi:10.1016/0952-8180(96)00043-8. PMID8695138.
^ McQuaid, KR.; Laine, L. (May 2008). “A systematic review and meta-analysis of randomized, controlled trials of moderate sedation for routine endoscopic procedures”. Gastrointest Endosc. 67 (6): 910–23. doi:10.1016/j.gie.2007.12.046. PMID18440381.
^ Article 4A of Export Control Order 2008 – provisions supplementing “the torture Regulation”
^ Riezzo I, Centini F, Neri M, Rossi G, Spanoudaki E, Turillazzi E, Fineschi V (2009). “Brugada-like EKG pattern and myocardial effects in a chronic propofol abuser”. Clin Toxicol. 47 (4): 358–63. doi:10.1080/15563650902887842. PMID19514884. S2CID22531823.
^ Kranioti EF, Mavroforou A, Mylonakis P, Michalodimitrakis M (22 March 2007). “Lethal self-administration of propofol (Diprivan): A case report and review of the literature”. Forensic Science International. 167 (1): 56–8. doi:10.1016/j.forsciint.2005.12.027. PMID16431058.
^ In Sweetman SC (Ed.). Martindale: The Complete Drug Reference 2005. 34th Edn London pp. 1305–7
^ Baudoin Z. General anesthetics and anesthetic gases. In Dukes MNG and Aronson JK (Eds.). Meyler’s Side Effects of Drugs 2000. 14th Edn Amsterdam pp. 330
^ Jump up to:ab Roussin A, Montastruc JL, Lapeyre-Mestre M (21 October 2007). “Pharmacological and clinical evidences on the potential for abuse and dependence of propofol: a review of the literature”. Fundamental and Clinical Pharmacology. 21 (5): 459–66. doi:10.1111/j.1472-8206.2007.00497.x. PMID17868199. S2CID22477291.
^ Langley, M; Heel, R (1988). “Propofol. A review of its pharmacodynamic and pharmacokinetic properties and use as an intravenous anaesthetic”. Drugs. 35 (4): 334–72. doi:10.2165/00003495-198835040-00002. PMID3292208.
^ Vesta, Kimi; Shaunta’ Martina; Ellen Kozlowski (25 April 2009). “Propofol-Induced Priapism, a Case Confirmed with Rechallenge”. The Annals of Pharmacotherapy. 40 (5): 980–982. doi:10.1345/aph.1G555. PMID16638914. S2CID36563320.
^ Fuentes, Ennio; Silvia Garcia; Manuel Garrido; Cristina Lorenzo; Jose Iglesias; Juan Sola (July 2009). “Successful treatment of propofol-induced priapism with distal glans to corporal cavernosal shunt”. Urology. 74 (1): 113–115. doi:10.1016/j.urology.2008.12.066. PMID19371930.
^ Eumorfia Kondili; Christina Alexopoulou; Nectaria Xirouchaki; Dimitris Georgopoulos (2012). “Effects of propofol on sleep quality in mechanically ventilated critically ill patients: a physiological study”. Intensive Care Medicine. 38 (10): 1640–1646. doi:10.1007/s00134-012-2623-z. PMID22752356. S2CID21206446.
^ Kim, MD, FACEP, Tae Eung; Shankel, MD, Tamara; Reibling, PhD, MA, Ellen T.; Paik, MSN, RN, Jacqueline; Wright, PhD, RN, Dolores; Buckman, PhD, RN, Michelle; Wild, MS, RN, Kathi; Ngo, MS, Ehren; Hayatshahi, PharmD, Alireza (1 January 2017). “Healthcare students interprofessional critical event/disaster response course”. American Journal of Disaster Medicine. 12 (1): 11–26. doi:10.5055/ajdm.2017.0254. ISSN1932-149X. PMID28822211.
^ Vasile B, Rasulo F, Candiani A, Latronico N (2003). “The pathophysiology of propofol infusion syndrome: a simple name for a complex syndrome”. Intensive Care Medicine. 29 (9): 1417–25. doi:10.1007/s00134-003-1905-x. PMID12904852. S2CID23932736.
^ Trapani, G; Latrofa, A; Franco, M; Altomare, C; Sanna, E; Usala, M; Biggio, G; Liso, G (1998). “Propofol analogues. Synthesis, relationships between structure and affinity at GABAA receptor in rat brain, and differential electrophysiological profile at recombinant human GABAA receptors”. Journal of Medicinal Chemistry. 41 (11): 1846–54. doi:10.1021/jm970681h. PMID9599235.
^ Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL (April 2001). “General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of gamma-aminobutyric acid (GABA) current at the GABA(A) receptor but not with lipid solubility”. J. Pharmacol. Exp. Ther. 297 (1): 338–51. PMID11259561.
^ Fowler CJ (February 2004). “Possible involvement of the endocannabinoid system in the actions of three clinically used drugs”. Trends Pharmacol. Sci. 25 (2): 59–61. doi:10.1016/j.tips.2003.12.001. PMID15106622.
^ Lee, U; Mashour, GA; Kim, S; Noh, GJ; Choi, BM (2009). “Propofol induction reduces the capacity for neural information integration: implications for the mechanism of consciousness and general anesthesia”. Conscious. Cogn. 18 (1): 56–64. doi:10.1016/j.concog.2008.10.005. PMID19054696. S2CID14699319.
^ Veselis RA, Reinsel RA, Feshchenko VA, Wroński M (October 1997). “The comparative amnestic effects of midazolam, propofol, thiopental, and fentanyl at equisedative concentrations”. Anesthesiology. 87 (4): 749–64. doi:10.1097/00000542-199710000-00007. PMID9357875. S2CID30185553.
^ James, R; Glen, JB (December 1980). “Synthesis, biological evaluation, and preliminary structure-activity considerations of a series of alkylphenols as intravenous anesthetic agents”. Journal of Medicinal Chemistry. 23 (12): 1350–1357. doi:10.1021/jm00186a013. ISSN0022-2623. PMID7452689.
^ Kvam C, Granese D, Flaibani A, Pollesello P, Paoletti S (1993). “Hyaluronan can be protected from free-radical depolymerization by 2, 6-diisopropylphenol, a novel radical scavenger”. Biochem. Biophys. Res. Commun. 193 (3): 927–33. doi:10.1006/bbrc.1993.1714. PMID8391811.
For the Reduction of The Severity and Incidence of Radiation and Chemotherapy-Induced Oral Mucositis
Avasopasem manganese, also known as GC4419, is a highly-selective small molecule mimetic of superoxide dismutase (SOD) being investigated for the reduction of radiation-induced severe oral mucositis.1,2 This drug has potential application for radiation-induced esophagitis and oral mucositis, in addition to being currently tested against COVID-19.
Avasopasem manganese is a superoxide dismutase mimetic that rapidly and selectively converts superoxide to hydrogen peroxide and oxygen in order to protect normal tissue from radiation therapy-induced damage.1 This drug is currently being investigated against oral mucositis, esophagitis, and COVID-19.
Transition metal pentaaza 15-membered macrocyclic ring complexes having the macrocyclic ring system corresponding to Formula A have been shown to be effective in a number of animal and cell models of human disease, as well as in treatment of conditions afflicting human patients.
For example, in a rodent model of colitis, one such compound, GC4403, has been reported when administered by intraperitoneal (ip) injection to significantly reduce the injury to the colon of rats subjected to an experimental model of colitis (see Cuzzocrea et al., Europ. J. Pharmacol., 432, 79-89 (2001)).
GC4403 administered ip has also been reported to attenuate the radiation damage arising both in a clinically relevant hamster model of acute, radiation-induced oral mucositis (Murphy et al., Clin. Can. Res., 74(13), 4292 (2008)), and lethal total body irradiation of adult mice (Thompson et al., Free Radical Res., 44(5), 529-40 (2010)).
Similarly, another such compound, GC4419, administered ip has been shown to attenuate VEGFr inhibitor-induced pulmonary disease in a rat model (Tuder, et al., Am. J. Respir. Cell Mol. Biol., 29, 88-97 (2003)), and to increase the anti-tumor activity of anti-metabolite and anti-mitotic agents in mouse cancer models (see, e.g., WO2009/143454). In other studies, GC4419 and GC4403 have been shown to be similarly potent in various animal models of disease. Additionally, another such compound, GC4401, administered ip has been shown to provide protective effects in animal models of septic shock (S. Cuzzocrea, et. al., Crit. Care Med., 32(1 ), 157 (2004)) and pancreatitis (S. Cuzzocrea, et. al., Shock, 22(3), 254-61 (2004)).
[0003] Certain of these compounds have also been shown to possess potent anti-inflammatory activity and prevent oxidative damage in vivo. For example, GC4403 administered ip has been reported to inhibit inflammation in a rat model of inflammation (Salvemini, et.al., Science, 286, 304 (1999)), and prevent joint disease in a rat model of collagen-induced arthritis (Salvemini et al., Arthritis & Rheumatism, 44(12), 2009-2021 (2001)). In addition, these compounds have been reported to possess analgesic activity and to reduce inflammation and edema by systemic administration in the rat-paw carrageenan hyperalgesia model, see, e.g., U.S. Pat. No. 6,180,620.
[0004] Compounds of the class comprising GC4419 have also been shown to be safe and effective in the prevention and treatment of disease in human subjects. For example, GC4419 administered by intravenous (iV) infusion has been shown to reduce oral mucositis in head-and-neck cancer patients undergoing chemoradiation therapy (Anderson, C, Phase 1 Trial of Superoxide Dismutase (SOD) Mimetic GC4419 to Reduce Chemoradiotherapy (CRT)-lnduced Mucositis (OM) in Patients (pts) with Mouth or Oropharyngeal Carcinoma (OCC), Oral Mucositis Research Workshop,
MASCC/ISOO Annual Meeting on Supportive Care in Cancer, Copenhagen, Denmark (June 25, 2015)).
[0005] However, the administered dose when delivered systemically, for example by a parenteral route, can be limited in animal models and particularly in humans by systemic exposure and resulting toxicity that appears to be similar in nature among the pentaaza 15-membered macrocyclic ring dismutase mimetics of Formula A, particularly GC4403, GC4419, GC4401 and related compounds sharing the dicyclohexyl and pyridine motif in the macrocycle ring (e.g., compounds sharing the dicyclohexyl and pyridine motif generally include compounds according to Formula (I) below herein having W as an unsubstituted pyridine moiety, and wherein U and V are transcyclohexanyl fused rings) . For example, the maximum tolerated dose of GC4403 delivered as a 30-minute iv infusion in humans is 25 mg, or roughly 0.35 mg/kg in a 70-kg subject, and similar limitations exist for animal parenteral dosing. Thus, the efficacy of treatment of conditions such as local inflammatory disease or tissue damage of the alimentary canal may be limited when using systemic delivery of GC4403 and similar compounds.
[0006] In each of these compounds comprising the pentaaza 15-membered macrocyclic ring of Formula A, the five nitrogens contained in the macrocyclic ring each form a coordinate covalent bond with the manganese (or other transition metal coordinated by the macrocycle) at the center of the molecule. Additionally, manganese (or other appropriate transition metal coordinated with the macrocycle) forms coordinate covalent bonds with “axial ligands” in positions perpendicular to the roughly planar macrocycle. Such coordinate covalent bonds are characterized by an available “free” electron pair on a ligand forming a bond to a transition metal via donation and sharing of the electron pair thus forming a two-electron bond between the metal and the donor atom of the ligand (Cotton, F.A. & G. Wilkinson, Advanced Inorganic Chemistry, Chapter 5, “Coordination Compounds”, 2nd revised edn., Interscience Publishers, p.139 (1966); lUPAC Gold Book, online version http://goldbook.iupac.org/C01329.html). The coordinate covalent nature of the bonds between manganese (or other such appropriate transition metal) and the five macrocyclic ring nitrogens and between manganese (or other such transition metal) and each of the two chloro axial ligands is evidenced, for example, by the “single crystal” X-ray crystal structure of GC4403 (Fig. 11 ) and GC4419 (Fig. 12).
[0007] Coordination compounds contrast with ionic compounds, for example, salts, where in the solid state the forces between anions and cations are strictly coulombic electrostatic forces of attraction between ions of opposite charge. Thus, in salts, discrete cations and anions provide the force to maintain the solid state structure; e.g., such as the chloride ion and the sodium ion in a typical salt such as sodium chloride (Cotton, F.A. & G. Wilkinson, Advanced Inorganic Chemistry, Chapter 5, “The Nature of Ionic Substances”, 2nd revised edn., Interscience Publishers, pp. 35-36, 45-49 (1966).
[0008] Although pentaaza 15-membered macrocyclic ring complexes have been disclosed in the literature for a number of anti-inflammatory indications, the representative disclosures discussed above illustrate that such compounds are generally administered by intraperitoneal (ip) or intravenous (iv) injection to potentiate systemic bioavailability. Local (e.g. topical) administration has been reported as ineffective in animal models of inflammatory disease, particularly when measured against the efficacy of systemic administration methods (Murphy et al., Clin. Can. Res., 74(13), 4292 (2008); WO 2008/045559). One research group has reported inhibition of colonic tissue injury and neutrophil accumulation by intracolonic administration of a prototype pentaaza macrocycle superoxide dismutase mimetic (MnPAM) (having a different structure from GC4403), though that disclosure neither addresses systemic bioavailability of the compounds described therein, nor explore limitations resulting from systemic bioavailability impacting safety and/or efficacy of that specific compound (Weiss et al., J. Biol. Chem., 271(42): 26149-26156 (1996); Weiss, R. and Riley, D., Drugs Future, 21 (4): 383-389 (1996)).
[0009] Aspects of the present disclosure provide for formulations of pentaaza macrocyclic ring complexes of the class comprising GC4419, GC4403, and GC4401 that exhibit limited systemic bioavailability when administered orally (e.g. less than 20%, less than 15%, and even less than 10% bioavailability when dosed in appropriate oil-based formulations; see Table 1 and when combined with other formulations even less than 5%, and even less than 1%; see Example 28). In general, drug absorption from the gastrointestinal tract occurs via passive uptake so that absorption is favored when the drug is in a non-ionized (neutral) and lipophilic form. See, e.g., Goodman & Gilman’s: The Pharmacological Basis of Therapeutics, Ninth Edition, p. 5-9 (1996). Without wishing to be limited to any particular theory, this is also believed to be the case for this class of compounds, as exemplified by GC4403, where the axial ligands are both chloro moieties forming a coordinate covalent bond to the manganese and a neutral complex results:
The Mn(ll) pentaaza macrocyclic ring dichloro complexes, such as GC4419, GC4401, GC4444, and GC4403 (structures shown below) were synthesized using literature procedures. For GC4403 the chiral R,R-diaminocyclohexane is utilized as starting material,2 whereas for GC4419, the mirror-image enantiomer of GC4403, the chiral S,S-diaminocyclohexane is utilized instead.3,4 The remainder of the synthesis of GC4419 can be identical in all respects to the method published for GC4403.2 The synthesis of the GC4401 complex was reported previously in reference 5.
[00213] The synthesis of GC4444 which contains the additional 11-R-Methyl substituent generating a fifth chiral center on carbon (and is also derived from R,R-diaminocyclohexane) is made from the corresponding chiral tetraamine whose synthesis was published in reference 6 as Example 5C.
Syntheses of Axial Ligand Derivatives
[00214] The same Mn(II) pentaaza macrocyclic ring dichloro complexes (GC4419, GC4403, GC4444 and GC4401 ) were also used as the starting material precursors for the syntheses of other axial ligand bound derivatives using a generic synthesis scheme in which a large excess of a salt of an anion is used to displace the chloro ligand thereby generating the new compound.
[00216] Using a 500-mL Erlenmeyer, 100 mL of deionized (“DI”) water was added to 5.3 g of GC4419; the mixture was stirred vigorously for 15-20 min, then sonicated for 5 min. The resulting light brownish suspension was filtered through a 10-20 μ fritted funnel (ca. 0.3 g of solid material remained in the funnel). The resulting clear solution was added into a sodium acetate solution (ca. 429 mmol, 21 equiv in 100 mL DI water) as a stream in one portion. No solid separated and the yellowish solution was stirred for 5 additional min. The solution was transferred to a separatory funnel and extracted (3 X 50 mL) with dichloromethane. The organic layers were separated, combined, and transferred back into a separatory funnel. The dichloromethane solution was back-extracted (2 X 50 mL) with aqueous sodium acetate (32 g/100 mL). The dichloromethane layer was dried over MgSO4 (ca. 10 g) for 30 min (w/stirring), filtered using a 10-20 μ fritted funnel, and the solution taken to dryness using a rotavap. To the yellow oily solid resulting from taking the solution to dryness was added methanol (50 mL). This solution was then again taken to dryness on the rotovap to yield a light yellow foam/glass. This material was dried in vacuo at room temperature for two days.
[00217] The isolated yellowish brittle (4.11 g, 75% yield based on GC4419) was analyzed by HPLC and showed a purity of 99.7% and elemental analysis showed 0.98 wt. % residual chlorine. The elemental analysis is consistent with the expected bis-(acetato) structure C25H41MnN5O4●2H2O. Anal Cal’d: C, 53.00% ; H, 8.01 %; N, 12.36%, and Mn, 9.70%. Anal Found: C, 53.10% ; H, 8.34% ; Mn, 9.86%, N, 12.56%, and CI (as total halogen content), 0.98 wt. %.
Patent
WO 2002071054
https://patents.google.com/patent/WO2002071054A1/enSuperoxide dismutase (SOD) enzymes are enzymes that catalyze the dismutation of the free radical superoxide, the one-electron reduction product of molecular oxygen. The dismutation of the free radical superoxide involves the conversion of this one-electron reduction product of molecular oxygen to the nonradical molecular oxygen. Superoxide dismutase enzymes are a class of oxidoreductases which contain either Cu/Zn, Fe, or Mn at the active site. Superoxide dismutase (SOD) mimetic compounds are low molecular weight catalysts which mimic the natural enzyme function of the superoxide dismutase enzymes. Thus, superoxide dismutase mimetic compounds also catalyze the conversion of superoxide into oxygen and hydrogen peroxide, rapidly eliminating the harmful biologically generated superoxide species that are believed to contribute to tissue pathology in a number of diseases and disorders. These diseases and disorders include reperfusion diseases, such as those following myocardial infarct or stroke, inflammatory disorders such as arthritis, and neurological disorders such as Parkinson’s disease. Chem Reviews, 1999 vol 99, No. 9, 2573-2587.Superoxide dismutase mimetic compounds possess several advantages over the superoxide dismutase enzymes themselves in that their chemical properties can be altered to enhance stability, activity and biodistribution while still possessing the ability to dismutase the harmful superoxide. Superoxide dismutase mimetic compounds have generated intense interest and have been the focus of considerable efforts to develop them as a therapeutic agent for the treatment of a wide range of diseases and disorders, including reperfusion injury, ischemic myocardium post-ischemic neuropathies, inflammation, organ transplantation and radiation induced injury. Most of the superoxide dismutase mimics currently being developed as therapeutic agents are synthetic low molecular weight manganese-based superoxide dismutase mimetic compounds. Chem Reviews, 2576. Superoxide dismutase mimetic compounds are metal complexes in which the metal can coordinate axial ligands. Examples of such metal complexes include, but are not limited to, complexes of the metals Mn and Fe. Many of the complexes of the metals Mn and Fe do not possess superoxide dismutase activity but possess properties that enable them to be put to other therapeutic and diagnostic uses. These therapeutic and diagnostic uses include MRI imaging enhancement agents, peroxynitrite decomposition catalysts, and catalase mimics. These metal complexes, however, share the structural similarity of possessing a metal that can coordinate exchangeable ligands. These metal complexes exist in water as a mixture of species in which various ligands are possible. An illustration of such a mixture is provided by M40403 , a Mn(π) complex of a nitrogen-containing fifteen membered macrocyclic ligand, shown in Scheme 1. One of the forms for this metal complex is the dichloro complex, which when dissolved in water another form is generated where one of the chloride anions immediately dissociates from the metal generating the [Mn(Cl)(aquo)]+ complex. The problem in aqueous solvent systems or any solvent which has a potential donor atom is that there are a variety of potential ligands available to coordinate axially to the Mn(π) ion of the complex, hi conducting an analysis of a sample containing a metal complex by high performance liquid chromatography (HPLC) the chromatogram tends to be very broad and unresolved due to the presence of the various species of complexes, as shown in Scheme 1. This phenomena makes the identification and quantification of metal complexes by standard HPLC techniques quite difficult. Therefore, in light of the developing roles of metal complexes as therapeutics in the treatment of various disorders and diagnostic agents, a substantial need exists for an effective and workable high performance liquid chromatography method for analyzing metal complexes.
Scheme 1An additional complication which exists is the issue of the acid stability of the metal complex. As the pH decreases, the rate at which the complex becomes protonated and experiences instability increases. This presents particular problems for the use of HPLC as a method of detection and quantification of the metal complexes because the mobile phase used for reverse phase HPLC frequently contains mixtures of organic solvents and water in various combinations with trifluoroacetic acid. The trifluoroacetic acid is commonly present between about 0.1 to about 0.5% by weight. The presence of the trifluoroacetic acid causes the complex to dissociate. This dissociation destroys the potential of any such method to be used for release testing for purity. Furthermore, the trifluoroacetate anion causes the formation of some of the trifluoroacetato complex which could possess a different retention time from the chloro complexes thus, confusing the chromatography. Thus, the phenomenon of ligand exchange, coupled with the acid instability of the metal complexes, provides considerable challenges to the effort to detect and quantify metal complexes using HPLC. These challenges and needs have surprisingly been met by the invention described below.Analytical HPLC is a powerful method to obtain information about a sample compound including information regarding identification, quantification and resolution of a compound. HPLC has been used particularly for the analysis of larger compounds and for the analysis of inorganic ions for which liquid chromatography is unsuitable. Skoog, D.A., West, M.A., Analytical Chemistry, 1986, p. 520. As an analytical tool HPLC takes advantage of the differences in affinity that a particular compound of interest has for the stationary phase and the mobile phase (the solvent being continuously applied to the column). Those compounds having stronger interactions with the mobile phase than with the stationary phase will elute from the column faster and thus have a shorter retention time. The mobile phase can be altered in order to manipulate the interactions of the target compound and the stationary phase. In normal-phase HPLC the stationary phase is polar, such as silica, and the mobile phase is a nonpolar solvent such as hexane or isopropyl ether. In reversed- phase HPLC the stationary phase is non-polar, often a hydrocarbon, and the mobile phase is a relatively polar solvent. Since 1974 when reversed-phase packing materials became commercially available, the number of applications for reversed- phase HPLC has grown, and reversed- phase HPLC is now the most widely used type of HPLC. Reversed-phase HPLC’s popularity can be attributed to its ability to separate a wide variety of organic compounds. Reversed-phase chromatography is especially useful in separating the related components of reaction mixtures, and therefore is a useful analytical tool for determining the various compounds produced by reactions. To create a non-polar stationary phase silica or synthetic polymer based adsorbents are modified with hydrocarbons. The most popular bonded phases are Cl, C4, C8 and C18. Silica based adsorbents modified with trimethylchlorosilane (Cl) and butyldimethylchlorosilane (C4) have a few applications in HPLC, mainly for protein separation or purification. These adsorbents show significant polar interactions. Octyl (C8) and octadecyl (C18) modified adsorbents are the most widely used silica based adsorbents, with almost 80% of all HPLC separations being developed with these adsorbents.The most important parameter in reversed-phase HPLC is the mobile phase. The type of mobile phase employed in the HPLC will have a significant effect on the retention of the analytes in the sample, and varying the composition of the mobile phase allows the chromatographer to adjust the retention times of target components in the mixture to desired values. This ability provides the HPLC method with flexibility. The mobile phase in reversed-phase chromatography has to be polar and it also has to provide reasonable competition for the adsorption sites for the analyte molecules. Solvents that are commonly employed as eluent components in reversed-phase HPLC are acetonitrile, dioxane, ethanol, methanol, isopropanol, tetrahydrofuran, and water. In reversed phase HPLC of high molecular weight biological compounds, the solvents acetonitrile, isopropanol or propanol are most frequently used. Popular additives to the mobile phase for the improvement of resolution include mixtures of phosphoric acid and amines and periϊuorinated carboxylic acids, especially trifluoroacetic acid (TFA). HPLC exploits the differences in affinity that a particular compound of interest has for the stationary phase and the mobile phase. This phenomenon can be utilized to separate compounds based on the differences in their physical properties. Thus, HPLC can be used to separate stereoisomers, diastereomers, enantiomers, mirror image stereoisomers, and impurities. Stereoisomers are those molecules which differ from each other only in the way their atoms are oriented in space. The particular arrangement of atoms that characterize a particular stereoisomer is known as its optical configuration, specified by known sequencing rules as, for example, either + or – (also D or L) and/or R or S. Stereoisomers are generally classified as two types, enantiomers or diastereomers. Enantiomers are stereoisomers which are mirror-images of each other. Enantiomers can be further classified as mirror-image stereoisomers that cannot be superimposed on each other and mirror-image stereoisomers that can be superimposed on each other. Mirror- image stereoisomers that can be superimposed on each other are known as meso compounds. Diastereomers are stereoisomers that are not mirror images of each other. Diastereomers have different physical properties such as melting points, boiling points, solubilities in a given solvent, densities, refractive indices, etc. Diastereomers can usually be readily separated from each other by conventional methods, such as fractional distillation, fractional crystallization, or chromatography, including HPLC.Enantiomers, however, present special challenges because their physical properties are identical. They generally cannot be separated by conventional methods, especially if they are in the form of a racemic mixture. Thus, they cannot be separated by fractional distillation because their boiling points are identical and they cannot be separated by fractional crystallization because their solubilites are identical (unless the solvent is optically active). They also cannot be separated by conventional chromatography such as HPLC because (unless the adsorbent is optically active) they are held equally onto the adsorbent. HPLC methods employing chiral stationary phases are a very common approach to the separation of enantiomers. To be able to separate racemic mixtures of stereoisomers, the chiral phase has to form a diastereomeric complex with one of the isomers, or has to have some other type of stereospecific interaction. The exact mechanism of chiral recognition is not yet completely understood. In reversed-phaseHPLC a common type of chiral bonded phase is chiral cavity phases.The ability to be able to separate diastereomers and enantiomers by HPLC is a useful ability in evaluating the success of synthetic schemes. It is often desirable to separate stereoisomers as a means of evaluating the enantiomeric purity of production samples. All references listed herein are hereby incorporated by reference in their entiretyExamples 1 (traditional mobile phase) and 2 (mobile phase containing excess of salt of a coordinating anion).
+X“
Scheme 2 Any metal complex possessing a metal that is capable of coordinating a monodentate ligand can be used in the present invention. Examples of such metal complexes include, but are not limited to, complexes of the metals Mn and Fe. The metal complexes of the invention preferably have therapeutic and diagnostic utilities. These therapeutic and diagnostic utilities include, but are not limited to, use as superoxide dismutase mimetic compounds, MRI imaging enhancement agents, peroxynitrite decomposition catalysts, and catalase mimics. The preferred metal complexes for use in the invention are superoxide dismutase mimetic compounds. Examples of such superoxide dismutase mimetic compounds include, but are not limited to, the following complexes of the metals Mn and Fe. Iron based superoxide dismutase mimetics include, but are not limited to, Fera(salen) complexes, Fera(l,4,7,10,13-pentaazacyclopentadecane) derivatives and Feffl(porphyrinato) complexes. Manganese based superoxide dismutase mimetic compounds include, but are not limited to, metal complexes containing manganese(π) or manganese(m). Examples of manganese based superoxide dismutase mimetic compounds include Mnm(porphyrinato) complexes, Mnffl(salen) complexes, and Mnπ(l ,4,7, 10, 13-pentaazacyclopentadecane) derivatives. Mnπ(l ,4,7, 10,13- pentaazacyclopentadecane) derivatives are more preferred for use in the invention. Examples of Mnπ(l,4,7,10,13-pentaazacyclopentadecane) derivatives preferred for use in the invention include, but are not limited to, M40403 and M40401, as shown in Scheme 3 below.Furthermore, stereoisomers of all of the above metal complexes can be used in the process of the present invention. Diastereomers of the same metal complexes can also be detected and separated by the method of the present invention. As it is often desirable to separate stereoisomers as a means of evaluating the chemical and optical purity of production samples, the metal complexes can also comprise products of a reaction stream. Enantiomers of any of the metal complexes referenced above can be used in the chiral HPLC method of the invention for the separation of enantiomers of a metal complex.
M40403 M40401
M40484Scheme 3The ligand is a coordinating anion that binds to the metal cation of the metal complex. The coordinating anion can serve as an axial ligand for a superoxide dismutase mimetic compound. Examples of such anions include, but are not limited to, chloride anions, thiocyanate anions, stearate anions, acetate anions, trifluoroacetate anions, carboxylate anions, formate anions, or azide anions. Preferred anions include chloride anions, thiocyanate anions, and formate anions. More preferred anions are chloride anions. The more preferred anions in the chiral HPLC embodiment of the invention are thiocyanate anions. When present in an excess, the thiocyanate anions bind to the coordinating metal of the complexes preferentially to the chloride anions. An excess of thiocyanate anions will produce the bis(thiocyanato) complexes of M40403 and M40419 as shown in Scheme 4.
M40403 M40403-(SCN)2
M40419 M40419-(SCN)2Scheme 4An example of the use of the acetate anion as the coordinating anion with M40403 is shown in Scheme 5 below. Scheme 6 illustrates the use of the formate anion as the coordinating anion with M40403.
M40403 M40403-(OAc)2Scheme 5
M40403 M40403-(Formate)2Scheme 6The coordinating anion is supplied by a salt of the coordinating anion. Salts of the chloride anion include, but are not limited to, sodium chloride, lithium chloride, potassium chloride, ammonium chloride, or tetraalkylammonium chloride. Preferred salts of the chloride anion include sodium chloride, lithium chloride and tetrabutylammonium chloride. Salts of the thiocyanate anion include, but are not limited to, sodium thiocyanate, potassium thiocyanate, ammonium thiocyanate, or lithium thiocyanate. Preferred salts of the thiocyanate anion include sodium thiocyanate and potassium thiocyanate. Salts of the acetate anion include, but are not limited to, potassium acetate, sodium acetate, ammonium acetate, ammonium trifluoroacetate and lithium acetate. Preferred salts of the acetate anion include ammonium acetate. Salts of the formate anion include, but are not limited to, potassium formate, sodium formate, ammonium formate and lithium formate. Preferred salts of the formate anion include ammonium formate. Salts of the cyanate anion include but are not limited to, sodium cyanate, potassium cyanate, or ammonium cyanate. Salts of the carboxylate anion include, but are not limited to, potassium carboxylate, ammonium carboxylate and sodium carboxylate. Salts of the stearate anion include, but are not limited to, lithium stearate and sodium stearate. Salts of the azide anion include, but are not limited to, sodium azide, potassium azide, and lithium azide. The salt added to the mobile phase can also be a mixture of any of these salts. Examples include a mixture of tetrabutylammonium chloride and lithium chloride.EXAMPLESExperimental For Examples 1-8 Chemicals, Solvents and MaterialsAll solvents used in the study were HPLC grade or equivalent. All chemicals were ACS reagent grade or equivalent.HPLC System and Data AnalysisThe HPLC chromatography was performed using a Gilson system (Model 306 pump, Model 155 UN-V detector, Model 215 liquid handler, Unipoint Software,Win98), a Narian system (Model 310 pump, Model 340 UN-N detector, Model 410 autosampler Star Workstation, Win98) or SSI system (Acuflow Series IN pump, Acutect 500 UV-N detector, Alcott Model 718 autosampler, HP Model 3395 integrator).Example 1HPLC Analysis of M40403 using Method 1
M40403 Method 1: Analytical Column: Waters YMC ODS-AQ S5 120A (4.6 x 50 mm); System A: 0.1% trifluoroacetic acid in H2O; System B: 0.08% trifluoroacetic acid in acetonitrile; Gradient: 10-50% system B over 10 min; Flow rate: 3ml/min; Detector wavelength: 265. Injected 20 μl of stock solution of M40403 prepared by dissolving 1 mg in 1 ml of water and diluting with 1 ml of system A. The HPLC chromatogram of M40403 using method 1 is shown in Figure 1. Example 2 HPLC Analysis of M40403 using Method 2Method 2: Analytical Column: Waters YMC 9DS-AQ S5 12θΛ (4.6 x 50 MM); System A: 0.5 N aqueous NaCl; System B: 1 :4 water/CH3CN; Gradient: 10-50% system B over 9 min; Flow rate: 3mL/min; Detector wavelength: 265 nm. Injected 20 μl of stock solution of M40403 prepared by dissolving 1 mg in 1 ml of system A. The HPLC chromatogram of M40403 using method 2 is shown in Figure 2.Example 3 HPLC Analysis of M40403 using Method 3Method 3: Analytical Column: Waters Symmetry Shield RP18, 5 μm, 250 x 4.6 mm;Mobile Phase: Acetonitrile: 0.125 M Tetrabutylammonium Chloride in water (pH 6.5), 5%: 95% H20(v/v); Flow rate: 1 mL/min; Detection wavelength: 265nm. Injected 20 μl of stock solution of M40403 prepared by dissolving 1 mg in 1 ml of mobile phase. The HPLC chromatogram of M40403 using method 3 is shown in Figure 3.The HPLC chromatogram of M40403 and related compounds using method 3 is shown in Figure 3a. Method 3 allows a separation of M40402 (bisimine of M40403), M40414 (monoimine of M40403) and M40475 (free ligand of M40403) (see chromatogram in Figure 3a).Example 4HPLC Analysis of M40403 using Method 4Method 4: Analytical Column: Waters Symmetry Shield RP18, 5 μm, 250 x 4.6 mm; Mobile Phase: Acetonitrile: 0.125 M Tetrabutylammonium Chloride and 0.5 M LiCl in water (pH 6.5), 5%: 95% H20 (v/v); Flow rate: lmL/min; Detection wavelength: 265 nm. Injected 20 μl of stock solution of M40403 prepared by dissolving 1 mg in 1 ml of system A. The HPLC chromatogram of M40403 using method 4 is shown in Figure 4.The HPLC chromatogram of M40403 and related compounds using method 4 is shown in Figure 4a. Method 4 allows a separation of M40402 (bisimine of M40403), M40414 (monoimine of M40403) and M40475 (free ligand of M40403) and all diastereomers of M40403 (see chromatogram in Figure 4a).Example 5 HPLC Analysis of M40401 using Method 1
M40401 Method 1: Analytical Column: Waters YMC ODS-AQ S5 120A (4.6 x 50 mm); System A: 0.1 % trifluoroacetic acid in H2O; System B: 0.08% trifluoroacetic acid in acetonitrile; Gradient: 10-50% system B over 10 min; Flow rate: 3ml/min; Detector wavelength: 265. Injected 20 μl of stock solution of M40401 prepared by dissolving 1 mg in 1 ml of water and diluting with 1 ml of system A. The HPLC chromatogram of M40401 using method 1 is shown in Figure 5.Example 6 HPLC with various NaCl concentrations:An HPLC was taken of M40401 with various concentrations of NaCl.Analytical Column: Waters YMC 9DS-AQ S5 120 A (4.6 x 50 mm);System A: (A) H2O (no NaCl) ; (B) 0.01 M NaCl in water; (C) 0.5 M NaCl in water;System B: acetonitrile; Gradient: 0-100% system B over 10 min; Flow: 3 ml/min;Detector wavelength: 265 nm. Injected 20 μl of stock solution of M40401 prepared by dissolving 1 mg in 1 ml of system A. The HPLC chromatogram of M40401 using various NaCl concentrations is shown in Figure 6. Example 7 HPLC Analysis of M40401 using Method 2Method 2: Analytical Column: Waters YMC ODS-AQ S5 12θΛ (4.6 x 50 MM); System A: 0.5 N aqueous NaCl; System B: 1 :4 water/CH3CN; Gradient 1 : 10-50% system B over 9 min; Flow rate: 3 mL/min; Detector wavelength: 265 nm. Injected 20 μl of stock solution of M40403 prepared by dissolving 1 mg in 1 ml of system A.The HPLC chromatogram of M40401 using method 2 is shown in Figure 7. Method 2 allows a separation of M40472 (bisimine of M40401), M40473 (monoimine of M40401), free ligand of M40403 and two isomers of M40401 (M40406, M40404).Example 8HPLC Analysis of M40401 using Method 3Method 3: Analytical Column: Waters Symmetry Shield RP18, 5 m, 250 4.6 mm; Mobile Phase: Acetonitrile: 0.125 M Tetrabutylammom‘um Chloride in H20 (pH 6.5), 5: 95%) H20 (v/v); Flow rate: lmL/min; Detection wavelength: 265 nm. The HPLC chromatogram of M40401 using method 3 is shown in Figure 8.Method 3 allows a separation of M40472 (bisimine of M40401), M40473 (monoimine of M40401), free ligand of M40403 and two isomers of M40401 (M40406, M40404).Example 9 HPLC Analysis of M40401 using Method 4Method 4: Analytical Column: Waters Symmetry Shield RP18, 5 μm, 250 x 4.6 mm;Mobile Phase: Acetonitrile: 0.125 M Tetrabutylammonium Chloride and 0.5 M LiCl in water (pH 6.5), 5: 95%> H2O (v/v); Flow rate: 1 mL/min; Detection wavelength: 265 nm; Injected 20 μl of stock solution of M40401 prepared by dissolving 1 mg in 1 ml of a mobile phase. The HPLC chromatogram of M40401 using method 4 is shown in Figure 9.The HPLC chromatogram of M40401 and related compounds using method 4 is shown in Figure 9a. Method 4 allows a separation of M40472 (bisimine of M40401), M40473 (monoimine of M40401), free ligand of M40403 and two isomers of M40401 (M40406, M40404). Example 10HPLC of M40403-(HCOO“)2 Using Formate AnionAn HPLC of M40403 employing the formate anion was taken. Analytical Column: Waters YMC 9DS-AQ S5 120 A (4.6 x 50 mm); System A: 0.025 M ammonium formate in water; System B: 1 : 4 = 0.125 M ammonium formate in water/ acetonitrile; Gradient: 0-100% system B over 10 min; Flow: 3 ml/min;Detector wavelength: 265 nm. Injected 20 μl of stock solution of M40403-(Formate)2 prepared by dissolving 1 mg in 1 ml of system A. The HPLC chromatogram of M40403-(HCOO“)2 is shown in Figure 10.Example 11 HPLC of M40403-(OAc)2 Using Acetate AnionAn HPLC of M40403 employing the acetate anion was taken.Analytical Column: Waters YMC 9DS-AQ S5 120 A (4.6 x 50 mm); System A: 0.025 M ammonium acetate in water; System B: 1: 4 = 0.125 M ammonium acetate in water/ acetonitrile; Gradient: 0-100% system B over 10 min; Flow: 3 ml/min;Detector wavelength: 265 nm. Injected 20 μl of stock solution of M40403-(OAc)2 prepared by dissolving 1 mg in 1 ml of system A. The HPLC chromatogram of M40403 -(OAc)2 is shown in Figure 11.Example 12An HPLC method to separate the diastereomers of superoxide dismutase mimetic compound M40403. Four stereoisomer mixtures were prepared (Part A) as shown in Schemes 5-9 and then separated (Part B) via reversed-phase high performance liquid chromatography. Part A: Synthesis of Stereoisomers Of M40403M40403 is synthesized from its single-isomer, tetra-amine precursor M40400 in the reaction shown in Scheme 7.
M40400 M40402
M40403Scheme 7The various stereoisomers of M40403 are synthesized from the various isomers of 1,2-diaminocyclohexane which provides the chiral carbon centers in M40403. The 1,2-diaminocyclohexane isomers used to prepare the R,R+R,S) M40403 stereoisomer mixture of Set 1 are shown in Scheme 6. Similarly, the 1,2-diaminocyclohexane isomers used to prepare the (R,R+S,S) M40403 stereoisomer mixture of Set 2 are shown in Scheme 7. The 1,2-diaminocyclohexane isomers used to prepare the (R,S+R,S) M40403 stereoisomer mixture of Set 3 are shown in Scheme 8. The 1,2- diaminocyclohexane isomers used to prepare the (S,S+R,S) M40403 stereoisomer mixture of Set 4 are shown in Scheme 9. As shown in Schemes 6-9 the M40403 diastereomers are prepared by template cyclization, followed by reduction with sodium borohydride.
Scheme 8
(S.S.S.S)Scheme 9
(S.R.R.S)Scheme 10
Scheme 11Table 1
Part B: Separation of Stereoisomer MixturesChemicals, Materials, and MethodsTetrabutylammonium chloride hydrate (98%, 34,585-7) was purchased from Aldrich Chemical Company. Sodium chloride (99.6%, S-9888) was purchased from Sigma Chemical Company. All other solvents (HPLC-grade unless otherwise indicated) and reagents were purchased from Fisher Scientific and were of the finest grade available. The SymmetryShield® RP18 column (4.6 mm x 250 mm, 5 μm particle size) and its corresponding guard column were purchased from Waters Corporation. Reversed-Phase HPLC ExperimentsPreparation of Standard SolutionsHPLC Mobile phased was an aqueous solution consisting of 0.125 M tetrabutylammonium chloride (TBAC) and 0.5 M LiCl, prepared by adding tetrabutylammonium chloride hydrate (36.99 g) and solid LiCl (21.2 g) to a 1 L volumetric flask, diluting to volume with Millipore water, and inverting the flask several times to obtain a homogeneous solution. The resulting solution was filtered through a 0.45 μm nylon filter prior to use. Mobile phase B was HPLC-grade acetonitrile. Samples of each diastereoisomer set for HPLC-UN analysis were prepared at concentrations of ~ 3.0 mg/mL in a 50:50 mixture of 0.5 M LiCl in MeOH:
SOLID STATE FORMS OF AVASOPASEM MANGANESE AND PROCESS FOR PREPARATION THEREOF
Avasopasem manganese (GC4419), has the following chemical structure:
[0003] Avasopasem manganese is a highly selective small molecule superoxide dismutase (SOD) mimetic which is being developed for the reduction of radiation-induced severe oral mucositis (SOM). The compound is described in U.S. Patent No. 8,263,568.
[0004] Polymorphism, the occurrence of different crystalline forms, is a property of some molecules and molecular complexes. A single molecule may give rise to a variety of polymorphs having distinct crystal structures and physical properties like melting point, thermal behaviors (e.g., measured by thermogravimetric analysis (“TGA”), or differential scanning calorimetry (“DSC”)), X-ray diffraction (XRD) pattern, infrared absorption fingerprint, and solid state (13C) NMR spectrum. One or more of these techniques may be used to distinguish different polymorphic forms of a compound.
[0005] Different salts and solid state forms (including solvated forms) of an active pharmaceutical ingredient may possess different properties. Such variations in the properties of different salts and solid state forms and solvates may provide a basis for improving formulation, for example, by facilitating better processing or handling characteristics, changing the
dissolution profile in a favorable direction, or improving stability (polymorph as well as chemical stability) and shelf-life. These variations in the properties of different salts and solid state forms may also offer improvements to the final dosage form, for instance, if they serve to improve bioavailability. Different salts and solid state forms and solvates of an active pharmaceutical ingredient may also give rise to a variety of polymorphs or crystalline forms, which may in turn provide additional opportunities to assess variations in the properties and characteristics of a solid active pharmaceutical ingredient.
[0006] Discovering new solid state forms and solvates of a pharmaceutical product may yield materials having desirable processing properties, such as ease of handling, ease of processing, storage stability, and ease of purification or as desirable intermediate crystal forms that facilitate conversion to other polymorphic forms. New solid state forms of a pharmaceutically useful compound can also provide an opportunity to improve the performance characteristics of a pharmaceutical product. It enlarges the repertoire of materials that a formulation scientist has available for formulation optimization, for example by providing a product with different properties, including a different crystal habit, higher crystallinity, or polymorphic stability, which may offer better processing or handling characteristics, improved dissolution profile, or improved shelf-life (chemical/physical stability). For at least these reasons, there is a need for additional solid state forms (including solvated forms) of Avasopasem manganese.
EXAMPLES
Preparation of starting materials
[00119] Avasopasem manganese can be prepared according to methods known from the literature, for example U.S. Patent No. 8,263,568. Alternatively, Avasopasem manganese can be prepared by the template method reported for the enantiomeric analogue GC4403, which has the formula:
GC4403 is disclosed in International Appl. No. WO 98/58636 (as compound SC-72325) and Riley, D.P, and Schall, O.F., Advances in Inorganic Chemistry (2007), 59, 233-263. Thus, GC4403 can be synthesized via the template route described in the literature using the chiral R,R-l,2-diamminocyclohexane [Salvemini, D., et ah, Science (1999), 286, 304-6 , and Aston, K, et al., Inorg. Chem. (2001), 40(8), 1779-89] Avasopasem manganese (GC4419) can be prepared by the same method except that the chiral R,R-l,2-diamminocyclohexane is replaced with S,S-1 ,2-diamminocyclohexane.
Example 1: Preparation of Avasopasem manganese Form AMI
[00120] Avasopasem manganese (0.1 grams) was dissolved in dichloromethane (0.5 ml) at 25-30°C in a test tube. The solution was filtered through 0.45 micron filter and the clear solution was subjected to slow solvent evaporation at 25°C by covering the tube with paraffin film with a pin hole. After, 2 days, the obtained solid was analyzed by XRD- Form AMI; as shown in Figure 1
GlobeNewswire: Galera Therapeutics Announces Avasopasem Manganese Improved Markers of Chronic Kidney Disease in Patients Receiving Cisplatin [Link]
444.84CN109134365 discloses an active compound or medicinal salt with multi-target effects of VEGFR1~3, fibroblast growth factor receptor 1~3, RET, Kit and PDGFR, and its chemical structure formula is as follows: Formula I:
Chemical name: 4-(2-Fluoro-3chloro-(cyclopropylaminocarbonyl)aminophenoxy)-7-methoxy-6-quinoline carboxamide, the drug name is fluvatinib. The compound has strong activity and provides a potential new treatment option for patients with tumors such as liver and kidney.
At 20-30°C, 4-chloro-7-methoxyquinoline-6-carboxamide (550.0 g) was added to the reaction kettle. At 20-30°C, DMSO (16.5L) was added to the reactor. At 20-30°C, 2-fluoro-3chloro-4-aminophenol was added to the reactor. At 20-35°C, sodium tert-butoxide (229g) was slowly added to the reaction kettle under stirring for 10-15 minutes. The reaction kettle was heated to 96°C (internal temperature) in 1.5 hours. The reaction was stirred at 96-100°C for 6.5 hours, and no 4-amino-3-chloro-2 fluorophenol remained. The reaction was cooled to 20-30°C. Under stirring, 23.1L of water was slowly added to the reaction solution. During the process, a dark brown solid was precipitated. Keep the internal temperature below 40°C. Stir at 30-40°C for 0.5 hour. Cool to 20-30°C and filter. At 20-30°C, the filter cake and 3.5L of water are added to the reactor. Stir for 0.5 hour at 20-30°C. filter. At 20-30°C, the filter cake and 4.0L of water are added to the reactor. Stir for 0.5 hour at 20-30°C. After filtering, the filter cake was dried in a vacuum dryer at 40°C for 18 hours (phosphorus pentoxide used as a desiccant, and the oil pump was vacuumed). The solid was pulverized to obtain 758 g of off-white solid and dried at 40° C. for 18 hours (phosphorus pentoxide was used as the desiccant, and the oil pump was vacuumed) to obtain Example 1A.LCMS(ESI)m/z:362.0[M+1] +1 H NMR (400MHz, DMSO-d 6 ) δppm 8.68 (br s, 2H), 7.82-7.96 (m, 1H), 7.67-7.82 (m, 1H), 7.46-7.59 (m, 1H), 7.12-7.26 (m, 1H), 6.67-6.80 (m, 1H), 6.43-6.58 (m, 1H), 5.84 (s, 2H), 4.04 (s, 3H).Example 1B
Example 1A (6.05g) was added to a three-necked flask containing NMP (60mL), pyridine (1.32g) and phenyl chloroformate (5.20g) were added to the reaction system, and the reaction system was at room temperature (25-30°C). ) After stirring for 1 hour, the reaction was complete. Cyclopropylamine (2.84g) was also added to the reaction system. The reaction solution was stirred at room temperature (25-30°C) for 0.5 hours. The reaction was completed. Add 20 mL of ethanol to the reaction system and stir. Tap water (500 mL) was added to the reaction system, a solid was precipitated, filtered, and the filter cake was spin-dried under reduced pressure to obtain a crude product (orange solid, 5.26 g); the crude product was passed through a chromatography column (DCM: MeOH = 20/1~10 /1) Purification to obtain the product (orange solid, 3.12 g), the product was added with 4 mL of absolute ethanol and stirred at room temperature for 18 hours, filtered, the filter cake was washed with 1 mL of ethanol, and dried under reduced pressure to obtain Example 1B. This compound is obtained by adding 1 equivalent of hydrochloric acid, sulfuric acid or methanesulfonic acid in acetone or ethanol solution to obtain the corresponding salt.LCMS(ESI)m/z:445.0[M+1] +1 H NMR (400MHz, DMSO-d 6 ) ppm 8.66-8.71 (m, 2H), 8.12-8.20 (m, 2H), 7.72-7.93 (m, 2H), 7.45 (t, J = 9.16 Hz, 1H) ,7.28(d,J=2.76Hz,1H),6.58(d,J=5.02Hz,1H),4.05(s,3H),2.56-2.64(m,1H),0.38-0.77(m,4H)Example 1
Example 1B (1.5g, 3.37mmol) was added to EtOH (45mL), the reaction temperature was raised to 60°C, at this temperature, CH 3 SO 3 H (324.07mg, 3.37mmol, 240.05μL) was added dropwise to the reaction In the solution, after the dripping is completed, the reaction solution is dissolved, and the temperature of the reaction solution is naturally cooled to 15-20°C under stirring, and the reaction solution is stirred at this temperature for 2 hours. A large amount of brown solid precipitated, filtered, and the filter cake was rinsed with absolute ethanol (5 mL), and the obtained filter cake was spin-dried under reduced pressure at 50° C. without purification, and Example 1 was obtained.LCMS(ESI)m/z:445.0[M+1] +1 H NMR(400MHz,DMSO-d 6 )δppm 9.02(d,J=6.53Hz,1H)8.72(s,1H)8.18-8.27(m,2H)7.87-8.03(m,2H)7.65(s,1H )7.53(t,J=9.03Hz,1H)7.32(br s,1H)7.11(d,J=6.27Hz,1H)4.08(s,3H)2.55-2.62(m,1H)2.35(s,3H) 0.34-0.75(m,4H)
PATENT
https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2021143954&tab=FULLTEXT&_cid=P12-KSZPW4-91508-1Example 1 Preparation of fluvatinib crystal form I Add the free base of fluvatinib of formula I (50mg, 112.40umol) to EtOH (2mL), stir at 15-20℃ for 12h, filter to obtain a filter cake, add the filter cake to 200mL acetone, stir at 15-20℃ After 12h, filter and spin-dry the filter cake under reduced pressure at 40°C to obtain fluvatinib solid. The result of XRPD detection is shown in Figure 1, named as the crystalline form I of fluvatinib, and the detection results of DSC and TGA are shown in Figure 2. And Figure 3. Example 2 Preparation of crystal form I of fluvatinib mesylate (also referred to herein as “fluvatinib mesylate”) The 4-[3-chloro-4-(cyclopropylaminocarbonylamino)-2-fluoro-phenoxy]-7-methoxy-quinoline-6-carboxamide i.e. fluvatinib (0.5g, 1.12mmol) was added to EtOH (10mL) solvent, heated to 55~60℃, and methanesulfonic acid (108.02mg, 1.12mmol, 80.02μL, 1eq) was added to the reaction flask under stirring at this temperature, and the reaction solution was dissolved. , The reaction solution was cooled to 20 ~ 30 ℃, stirred at this temperature for 1 h, a brown solid precipitated out under vacuum filtration, the filter cake was rinsed with ethanol (2mL*2), and the filter cake was spin-dried at 40 ~ 50 ℃ under reduced pressure. The solid product, named as the crystalline form I of fluvatinib mesylate, was tested by XRPD, DSC, and TGA. The XRPD test results are shown in Table 1 and Figure 4 below, and the DSC and TGA test results are shown in Figure 5. Melting point is about 232-237°C.
Frontpro is currently authorised as chewable tablets for use in dogs. The variation concerns the change of legal status from prescription-only to non-prescription veterinary medicine. Additionally, the applicant is adding the list of local representatives to the package leaflet.
A particularly active isoxazoline compound, 4-[5-[3-chloro-5-(trifluoromethyl)phenyl]-4,5-dihydro-5-(trifluoromethyl)-3-isoxazolyl]-N-[2-oxo-2-[(2,2,24rifluoroethyl)amino]ethyl]-l-naphthalenecarboxamide, is known by the nonproprietary name afoxolaner. Afoxolaner has the following chemical structure:
Afoxolaner
Other isoxazoline compounds that have been found to be highly active against parasitic insects and arachnids are known by the nonproprietary names fluralaner (see US 7,662,972, which is incorporated herein by reference), sarolaner (see US 8,466, 15, incorporated herein by reference) and lotilaner (see, for example US 8,383,659, incorporated herein by reference). The structures of these compounds are shown below:
In addition, published patent application nos. US 2010/0254960 Al, WO 2007/070606
A2, WO 2007/123855 A2, WO 2010/003923 Al, US7951828 & US7662972, US 2010/0137372 Al, US 2010/0179194 A2, US 2011/0086886 A2, US 2011/0059988 Al, US 2010/0179195 Al and WO 2007/075459 A2 and U.S. Patent No. 7,951,828 (all incorporated herein by reference) describe various other parasiticidal isoxazoline compounds.
It is known in the field that isoxazoline compounds having a chiral quaternary carbon atom such as the carbon atom adjacent to the oxygen on the isoxazoline ring of the compounds described above have at least two optical isomer (enantiomers) that are mirror images of each other. Furthermore, it is sometimes the case with biologically active compounds that one of the enantiomers is more active than the other enantiomer. In addition, it is sometimes the case that one enantiomer of a biologically active compound is less toxic than the other enantiomer.
Therefore, with optically active compounds it is desirable to utilize the enantiomer that is most active and less toxic (eutomer). However, isolating the most active enantiomer from a mixture can be costly and result in waste of up to half of the racemic mixture prepared.
Processes to prepare certain isoxazoline compounds enriched in an enantiomer using some cinchona alkaloid-derived phase transfer catalysts have been described. For example, US 2014/0206633 Al, US 2014/0350261 Al, WO 2013/116236 Al and WO 2014/081800 Al (incorporated herein by reference) describe the synthesis of certain isoxazoline active agents enriched in an enantiomer using cinchona alkaloid-based chiral phase transfer catalysts. Further, Matoba et al., Angew. Chem. 2010, 122, 5898-5902 describes the chiral synthesis of certain pesticidal isoxazoline active agents. However, these documents do not describe the processes and certain catalysts described herein.
Scheme 3
Example 7: Preparation of (S)-afoxolaner using chiral phase transfer catalyst (Ilia- 13-1):
(ΠΑ-1) (^-afoxolaner
1) Starting material (IIA-1) (200g, 1.Oeq, 94.0%) and DCM (6 L, 30 volumes) were placed into a 10 L reactor, the solid was dissolved completely.
2) The mixture was cooled to 0°C, and some starting material precipitated out.
3) The catalyst (Ilia- 13-1) (7.56g, 3% mol, 95.0%) was added to the mixture and the resulting mixture cooled further to -10° C.
4) Hydroxylamine (64.9 g, 3.0 eq, 50% solution in water) was added to a solution of NaOH (52.5g, 4. Oeq, in 5v water) in a separate reactor and stirred for 30 minutes.
5) The resulting hydroxylamine/NaOH solution was then added dropwise to the 10 L reactor containing (IIA-1) over about 4 hours.
6) The resulting mixture was stirred for 12 hours at -10°C and monitored for the extent of reaction until the amount of starting material was < 1.0% by HPLC.
7) The mixture was then warmed to 10°C, 1 liter of water was added and the mixture was stirred for 10 minutes.
8) The mixture was allowed to settle to separate the two phases, and the organic layer was collected.
9) The organic layer was then washed with 2 liters of water, the layers were allowed to separate again and the organic layer was collected.
10) The organic layer was washed with 1 liter of brine, the layers allowed to separate and the organic layer was collected and dried over Na2S04 (200 g).
11) The dried organic layer was concentrated under vacuum to about 2 volumes.
12) Toluene (2 L, 10 volumes) was charged to the concentrated mixture and concentration under vacuum was continued to about 5 volumes. Solvent exchange was repeated twice again.
13) The resulting solution was placed into a 2.0 L reactor and heated to 55-60°C.
14) Cyclohexane (300 ml, 1.5 volumes) was added at 55-60°C.
15) The mixture was then cooled to 40 °C over 1.5 hours and then stirred at 40°C for 3 hours.
16) The mixture was then cooled to 25 °C over 2 hours and stirred at 25°C for a further 3 hours.
17) The resulting mixture was cooled to 0-5 °C over 1 hour and stirred at 5 °C for 12 hours, at which time the mixture was filtered to isolate the product.
18) The filter cake was washed with cold toluene/ Cyclohexane (3 : 1, 1000 ml, 5 volumes).
19) The product was obtained as a white solid. (171.5g, chiral purity > 99.0% by area using the chiral HPLC method described in Example 3, chemical purity > 99.0% by area (HPLC), yield: 83.6%, assay purity: 92%). The 1H NMR and LCMS spectra are consistent with the structure of (^-afoxolaner as the toluene solvate. Figure 3 shows the 1H NMR spectra of (S)-afoxolaner in DMSO-d6 and Figure 4 shows the 1H NMR spectra of afoxolaner (racemic) for comparison. The chiral purity of the product was determined using the chiral HPLC method described in Example 3. Figure 5 shows the chiral HPLC chromatogram of afoxolaner (racemic) and Figure 6 shows the chiral HPLC chromatogram of the product (^-afoxolaner showing one enantiomer.
Example 8: Alternate Process to prepare (^-afoxolaner
An alternate process for the preparation of (S)-afoxolaner was conducted. Some of the key variations in the alternate process are noted below.
1. 1 kilogram of compound (IIA-1) (1 eq.) and 9 liters of DCM are charged to a reactor and stirred to dissolve the compound.
2. The mixture is cooled to about 0° C and 50 grams (5 mole %) of the chiral phase transfer catalyst (Ilia- 13-1) and 1 liter of DCM are charged and the resulting mixture is cooled to about -13° C.
3. A solution of 19% (w/w) hydroxylamine sulfate (294 g, 1.1 eq.) (made with 294 grams of ( H2OH)H2S04 and 141 grams of NaCl in 1112 mL of water) and 4.4 equivalents of NaOH as a 17.6% (w/w) solution (286 grams NaOH and 158 grams of NaCl in 1180 mL water) are charged to the reaction mixture simultaneously.
4. The resulting reaction mixture was aged about 20 hours at about -13° C and then checked for reaction conversion by HPLC (target < 0.5% by area);
5. After completion of the reaction, water (3 vol.) was added at about 0° C. Then, a solution of 709 g of KH2P04 in 4.2 liters of water are added to the mixture to adjust the pH (target 7-8) and the resulting mixture is stirred at about 20° C for 30 minutes.
6. The layers are allowed to settle, the aqueous layer is removed and the organic layer is washed with 3 liters of water twice.
Crystallization of Toluene Solvate
1. After the extraction/washing step, the dichloromethane is removed by distillation under vacuum to about 1-2 volumes and toluene (about 5-10 volumes) is added.
2. The volume is adjusted by further distillation under vacuum and/or addition of more toluene to about 5-6 volumes. The mixture is distilled further while maintaining the volume to completely remove the dichloromethane reaction solvent.
3. The mixture is then cooled to about 10° C and seeded with afoxolaner (racemic compound) and stirred at the same temperature for at least 2 hours;
4. The mixture is heated to about 55-65° C, aged for at least 17 hours and then the solid is filtered off. The filtered solid is washed with toluene;
5. The combined filtrate and wash is adjusted to a volume of about 5-6 volumes by
distillation under vacuum and/or toluene addition;
6. The resulting mixture is cooled to about 10° C and aged for at least 5 hours then filtered.
The cake is washed with toluene.
7. The cake is dried at 50° C under vacuum to obtain a toluene solvate of (S)-afoxolaner containing between about 6% and 8% toluene.
Re-crystallization from cyclohexane/ethanol
The toluene solvate of (S)-afoxolaner was subsequently re-crystallized from a mixture of cyclohexane and ethanol to remove the associated toluene and to further purify the product.
1. 591 grams of the (S)-afoxolaner toluene solvate were charged to a vessel along with 709 mL of ethanol (1.2 vol.) and 1773 mL of cyclohexane (3 vol.) and the mixture heated to about 60° C.
2. To the resulting mixture was added an additional 6383 mL of cyclohexane with stirring.
3. The resulting mixture was cooled to about 30° C and then heated again to 60° C. This process was repeated once.
4. The mixture was slowly cooled to 10° C and stirred for at least 5 hours.
5. The resulting slurry was filtered and the cake washed with cyclohexane.
6. The cake was dried at 50° C under vacuum to provide 453.7 grams of (S)-afoxolaner
Example 9: Comparative selectivity of benzyloxy-substituted chiral phase transfer catalyst (Illa-13) with other cinchona alkaloid-based chiral phase transfer catalysts.
The selectivity of the formation of (S)-afoxolaner from compound IIA-1 as shown above was studied with sixteen chiral phase transfer catalysts (PTC) of different structures. The reaction was conducted using conditions similar to those of example 7. The ratio of (^-afoxolaner and (R)-afoxolaner in the reaction mixture was determined by chiral HPLC using the method described in Example 3. The results of the study are provided in Table 2 below.
Table 2
No. Chiral PTC Ratio of (S)- to (R)-afoxolaner
16 50% : 50%
As shown in the table, the catalyst in which the group R in the structure of formula (Ilia) is 3,4,5-tribenzyloxy phenyl results in a surprising improved selectivity for the (S)-enantiomer compared with other quinine-based phase transfer catalysts in which the group corresponding to R in formula (Ilia) is another group.
Example 10: Improvement of Chiral Purity of (<S)-afoxolaner by Crystallization from Toluene
A sample of reaction mixture containing a ratio (HPLC area) of 92.1 :7.9, (^-afoxolaner to (R)-afoxolaner, was concentrated to dryness and the residue was crystallized from toluene and from ethanol/cyclohexane using a process similar to that described in Example 8. The isolated crystalline solid was analyzed by chiral HPLC to determine the relative amounts of (S)-afoxolaner and (R)-afoxolaner (HPLC method: column – Chiralpak AD-3 150 mm x 4.6 mm x 3.0 μηι, injection volume – 10 μΐ., temperature – 35° C, flow – 0.8 mL/minute, mobile phase -89% hexane/10% isopropanol/1% methanol, detection – 312 nm). The ratio of (^-afoxolaner to (R)-afoxolaner in the solid isolated from the toluene crystallization was found to be 99.0 : 1.0 while the ratio of (S)-afoxolaner to (R)-afoxolaner in the solid crystallized from ethanol/cyclohexane was found to be 95.0 : 5.0.
The example shows that the crystallization (^-afoxolaner from an aromatic solvent such as toluene results in a significant improvement of chiral purity of the product. This is very unexpected and surprising.
Example 1 1 : Comparative selectivity of benzyl oxy vs. alkoxy-substituted chiral phase transfer catalyst of Formula (Ilia- 13)
Three chiral phase transfer catalysts of Formula (IIIa-13), wherein the phenyl ring is substituted with three alkoxy groups and three benzyloxy groups (R = methyl, ethyl and benzyl); R’=OMe, W=vinyl and X=chloro were evaluated in the process to prepare of (,S)-IA from compound IIA-1
as shown below.
The amount of solvents and reagents and the reaction and isolation conditions were as described in Example 7 above. The same procedure was used for each catalyst tested. It was found that the selectivity of the tri-benzyloxy catalyst was surprisingly significantly better than the two alkoxy-substituted catalysts, as shown by the chiral purity of the product. Furthermore, it was found that using the tri-benzyloxy substituted phase transfer catalyst the resulting chemical purity was also much better. The superior selectivity of the benzyloxy-substituted catalyst is significant and surprising and cannot be predicted. Chiral phase transfer catalysts containing a phenyl substituted with benzyloxy and alkoxy groups were found to be superior to catalysts substituted with other groups such as electron-withdrawing groups and alkyl groups. The chiral purity and chemical purity of the product produced from the respective phase-transfer catalysts is shown in the Table 3 below:
Afoxolaner is the active principle of the veterinary medicinal products NexGard (alone) and Nexgard Spectra (in combination with milbemycin oxime).[4][5][6] They are indicated for the treatment and prevention of flea infestations, and the treatment and control of tick infestations in dogs and puppies (8 weeks of age and older, weighing 4 pounds (~1.8 kilograms) of body weight or greater) for one month.[7] These products are administered orally and poisons fleas once they start feeding.
Afoxolaner is recommended to be administered at a dose of 2.7–7 mg/kg dog’s body weight.[11]
Toxicity for mammals
According to clinical studies performed prior marketing:
The oral toxicity profile of afoxolaner consists of a diuretic effect (rats only), effects secondary to a reduction in food consumption (rats and rabbits only) and occasional vomiting and/or diarrhoea (dogs, 120 and 200 mg/kg bodyweight (bw)) following high oral doses. No treatment-related effects on vomiting or diarrhoea were noted following oral doses of up to 31.5 mg/kg bw in the pivotal target animal safety study, nor in the EU field trial.[9]
Extralabel use of afoxolaner in a pet pig has been described without any adverse effects.[13] Experimental use in commercial pigs also did not result in any adverse effects.[14]
Shoop WL, Hartline EJ, Gould BR, Waddell ME, McDowell RG, Kinney JB, Lahm GP, Long JK, Xu M, Wagerle T, Jones GS, Dietrich RF, Cordova D, Schroeder ME, Rhoades DF, Benner EA, Confalone PN: Discovery and mode of action of afoxolaner, a new isoxazoline parasiticide for dogs. Vet Parasitol. 2014 Apr 2;201(3-4):179-89. doi: 10.1016/j.vetpar.2014.02.020. Epub 2014 Mar 14. [Article]
^ Casida JE (April 2015). “Golden age of RyR and GABA-R diamide and isoxazoline insecticides: common genesis, serendipity, surprises, selectivity, and safety”. Chemical Research in Toxicology. 28 (4): 560–6. doi:10.1021/tx500520w. PMID25688713.
VCH-759 had been in phase II clinical trials by ViroChem Pharma (acquired by Vertex in 2009) for the treatment of HCV infection. However, this research has been discontinued.
Infection with HCV is a major cause of human liver disease throughout the world. In the US, an estimated 4.5 million Americans are chronically infected with HCV. Although only 30% of acute infections are symptomatic, greater than 85% of infected individuals develop chronic, persistent infection. Treatment costs for HCV infection have been estimated at $5.46 billion for the US in 1997. Worldwide over 200 million people are estimated to be infected chronically. HCV infection is responsible for 40-60% of all chronic liver disease and 30% of all liver transplants. Chronic HCV infection accounts for 30% of all cirrhosis, end-stage liver disease, and liver cancer in the U.S. The CDC estimates that the number of deaths due to HCV will minimally increase to 38,000/year by the year 2010.
Due to the high degree of variability in the viral surface antigens, existence of multiple viral genotypes, and demonstrated specificity of immunity, the development of a successful vaccine in the near future is unlikely. Alpha-interferon (alone or in combination with ribavirin) has been widely used since its approval for treatment of chronic HCV infection. However, adverse side effects are commonly associated with this treatment: flu-like symptoms, leukopenia, thrombocytopenia, depression from interferon, as well as anemia induced by ribavirin (Lindsay, K. L. (1997) Hepatology 26 (suppl 1 ): 71 S-77S). This therapy remains less effective against infections caused by HCV genotype 1 (which constitutes -75% of all HCV infections in the developed markets) compared to infections caused by the other 5 major HCV genotypes. Unfortunately, only -50-80% of the patients respond to this treatment (measured by a reduction in serum HCV RNA levels and normalization of liver enzymes) and, of responders, 50-70% relapse within 6 months of cessation of treatment. Recently, with the introduction of pegylated interferon (Peg-IFN), both initial and sustained response rates have improved substantially, and combination treatment of Peg-IFN with ribavirin constitutes the gold standard for therapy. However, the side effects associated with combination therapy and the impaired response in patients with genotype 1 present opportunities for improvement in the management of this disease.
First identified by molecular cloning in 1989 (Choo, Q-L et al (1989) Science 244:359-362), HCV is now widely accepted as the most common causative agent of post-transfusion non A, non-B hepatitis (NANBH) (Kuo, G et al (1989) Science 244:362-364). Due to its genome structure and sequence homology, this virus was assigned as a new genus in the Flaviviridae family. Like the other members of the Flaviviridae, such as flaviviruses (e.g. yellow fever virus and Dengue virus types 1-4) and pestiviruses (e.g. bovine viral diarrhea virus, border disease virus, and classic swine fever virus) (Choo, Q-L et al (1989) Science 244:359-362; Miller, R.H. and R.H. Purcell (1990) Proc. Natl. Acad. Sci. USA 87:2057-2061 ), HCV is an enveloped virus containing a single strand RNA molecule of positive polarity. The HCV genome is approximately 9.6 kilobases (kb) with a long, highly conserved, noncapped 5′ nontranslated region (NTR) of approximately 340 bases which functions as an internal ribosome entry site (IRES) (Wang CY et al ‘An RNA pseudoknot is an essential structural element of the internal ribosome entry site located within the hepatitis C virus 5′ noncoding region’ RNA- A Publication of the RNA Society. 1 (5): 526-537, 1995 JuL). This element is followed by a region which encodes a single long open reading frame (ORF) encoding a polypeptide of -3000 amino acids comprising both the structural and nonstructural viral proteins.
Upon entry into the cytoplasm of the cell, this RNA is directly translated into a polypeptide of -3000 amino acids comprising both the structural and nonstructural viral proteins. This large polypeptide is subsequently processed into the individual structural and nonstructural proteins by a combination of host and virally-encoded proteinases (Rice, CM. (1996) in B.N. Fields, D.M.Knipe and P.M. Howley (eds) Virology 2nd Edition, p931-960; Raven Press, N.Y.). Following the termination codon at the end of the long ORF, there is a 3′ NTR which roughly consists of three regions: an – 40 base region which is poorly conserved among various genotypes, a variable length poly(U)/polypyrimidine tract, and a highly conserved 98 base element also called the “3′ X-tail” (Kolykhalov, A. et al (1996) J. Virology 70:3363-3371 ; Tanaka, T. et al (1995) Biochem Biophys. Res. Commun. 215:744-749; Tanaka, T. et al (1996) J. Virology 70:3307-3312; Yamada, N. et al (1996) Virology 223:255-261 ). The 3′ NTR is predicted to form a stable secondary structure which is essential for HCV growth in chimps and is believed to function in the initiation and regulation of viral RNA replication.
The NS5B protein (591 amino acids, 65 kDa) of HCV (Behrens, S. E. et al (1996) EMBO J. 15:12-22), encodes an RNA-dependent RNA polymerase (RdRp) activity and contains canonical motifs present in other RNA viral polymerases. The NS5B protein is fairly well conserved both intra-typically (-95-98% amino acid (aa) identity across 1 b isolates) and inter-typically (-85% aa identity between genotype 1 a and 1 b isolates). The essentiality of the HCV NS5B RdRp activity for the generation of infectious progeny virions has been formally proven in chimpanzees (A. A. Kolykhalov et al.. (2000) Journal of Virology, 74(4): 2046-2051 ). Thus, inhibition of NS5B RdRp activity (inhibition of RNA replication) is predicted to be useful to treat HCV infection.
Although the predominant HCV genotype worldwide is genotype 1, this itself has two main subtypes, denoted 1a and 1 b. As seen from entries into the Los Alamos HCV database
(www.hcv.lanl.gov) (Table 1 ) there are regional differences in the distribution of these subtypes: while genotype 1 a is most abundant in the United States, the majority of sequences in Europe and Japan are from genotype 1 b.
Table 1
Based on the foregoing, there exists a significant need to identify synthetic or biological compounds for their ability to inhibit replication of both genotype 1 a and genotype 1 b of HCV.
Compound A 5-Phenyl-3-[[(frans-4-methylcyclohexyl)carbonyl](1-methylethyl)amino]-2-thiophenecarboxylic acid
To a mixture of methyl S-^trans^-methylcyclohexyOcarbonylKI-methylethy^aminol-S-phenyl-2-thiophenecarboxylate (Intermediate 31 ) (390 mg) in THF/MeOH/water (3:2:1, vol/vol, 40 ml. total) was added lithium hydroxide monohydrate (246 mg). The mixture was stirred at room temperature for 20 hours, the solvents removed in vacuo, and the residue partitioned between water (40 ml.) and ethyl acetate (40 ml_). The organic layer was dried
(Na2SC>4), evaporated and triturated with ether to give the title compound. MS calcd for (C22H27NO3S+ H)+: 356 MS found (electrospray): (M+H)+ =356
Compounds A, B, C and D may be made according to the processes described in WO2002/100851 or as described hereinabove.
Structures of Compounds A, B, C and D are shown below for the avoidance of doubt.
The compounds of Formula (I) which have been tested demonstrate a surprisingly superior potency as HCV polymerase inhibitors, as shown by the IC5O values in the cell-based assays across both of the 1 a and 1 b genotypes of HCV, compared to Compounds A, B, C and D. Accordingly, the compounds of Formula (I) are of great potential therapeutic benefit in the treatment and prophylaxis of HCV.
REACH (EC 1907/2006)aims to improve the protection of human health and the environment through the better and earlier identification of the intrinsic properties of chemical substances. This is done by the four processes of REACH, namely the registration, evaluation, authorisation and restriction of chemicals. REACH also aims to enhance innovationand competitiveness of the EU chemicals industry.
“No data no market”: the REACH Regulation places responsibility on industry to manage the risks from chemicals and to provide safety information on the substances. Manufacturers and importers are required to gather information on the properties of their chemical substances, which will allow their safe handling, and to register the information in a central database in theEuropean Chemicals Agency (ECHA)in Helsinki. The Agency is the central point in the REACH system: it manages the databases necessary to operate the system, co-ordinates the in-depth evaluation of suspicious chemicals and is…
CAS Registry Number: 7491-74-9 CAS Name: 2-Oxo-1-pyrrolidineacetamide Additional Names: 2-pyrrolidoneacetamide; 2-pyrrolidinoneacetamide; 2-ketopyrrolidine-1-ylacetamide; 1-acetamido-2-pyrrolidinone Manufacturers’ Codes: UCB-6215 Trademarks: Avigilen (Riemser); Axonyl (Pfizer); Cerebroforte (Azupharma); Encetrop (Alpharma); Gabacet (Sanofi-Synthelabo); Geram (UCB); Nootrop (UCB); Nootropil (UCB); Nootropyl (UCB); Norzetam (UCB); Normabraïn (UCB); Piracebral (Hexal); Piracetrop (Holsten); Sinapsan (Rodleben)Molecular Formula: C6H10N2O2 Molecular Weight: 142.16 Percent Composition: C 50.69%, H 7.09%, N 19.71%, O 22.51% Literature References: Prepn: H. Morren, NL6509994; eidem,US3459738 (1966, 1969 both to U.C.B.). Pharmacology: Giurgea et al.,Arch. Int. Pharmacodyn. Ther.166, 238 (1967); Giurgea, Moyersoons, ibid.188, 401 (1970); Giurgea et al.,Psychopharmacologia20, 160 (1971). Metabolism and biochemical studies: Gobert, J. Pharm. Belg.27, 281 (1972). Clinical studies: W. J. Oosterveld, Arzneim.-Forsch.30, 1947 (1980); G. Chouinard et al.,Psychopharmacol. Bull.17, 129 (1981); in dyslexia: M. Di Ianni et al.,J. Clin. Psychopharmacol.5, 272 (1985).Properties: Crystals from isopropanol, mp 151.5-152.5°. Melting point: mp 151.5-152.5° Therap-Cat: Nootropic. Keywords: Nootropic.
Piracetam is in the racetams group, with chemical name 2-oxo-1-pyrrolidine acetamide. It is a derivative of the neurotransmitter GABA[5] and shares the same 2-oxo-pyrrolidone base structure with pyroglutamic acid. Piracetam is a cyclic derivative of GABA (gamma-aminobutyric acid). Related drugs include the anticonvulsants levetiracetam and brivaracetam, and the putative nootropics aniracetam and phenylpiracetam.Piracetam is a drug marketed as a treatment for myoclonus[3] and a cognitive enhancer.[4] Evidence to support its use is unclear, with some studies showing modest benefits in specific populations and others showing minimal or no benefit.[5][6] Piracetam is sold as a medication in many European countries. Sale of piracetam is not illegal in the United States, although it is not regulated nor approved by the FDA so it must be marketed as a dietary supplement.[4]
A 2001 Cochrane review concluded that there was not enough evidence to support piracetam for dementia or cognitive problems.[6] A 2005 review found some evidence of benefit in older subjects with cognitive impairment.[5] In 2008, a working group of the British Academy of Medical Sciences noted that many of the trials of piracetam for dementia were flawed.[7]
There is no good evidence that piracetam is of benefit in treating vascular dementia.[8]
Depression and anxiety
Some sources suggest that piracetam’s overall effect on lowering depression and anxiety is higher than on improving memory.[9] However, depression is reported to be an occasional adverse effect of piracetam.[10]
Peripheral vascular effects of piracetam have suggested its use potential for vertigo, dyslexia, Raynaud’s phenomenon and sickle cell anemia.[5][3] There is no evidence to support piracetam’s use in sickle cell crisis prevention[11] or for fetal distress during childbirth.[12] There is no evidence for benefit of piracetam with acute ischemic stroke,[13] though there is debate as to its utility during stroke rehabilitation.[14][15]
According to a 2005 review, piracetam has been observed to have the following side effects: hyperkinesia, weight gain, nervousness, somnolence, depression and asthenia.[5]
Piracetam reduces platelet aggregation as well as fibrinogen concentration, and thus is contraindicated to patients suffering from cerebral hemorrhage.[5][3]
Toxicity
Piracetam does not appear to be acutely toxic at the doses used in human studies.[6][18][19]
The LD50 for oral consumption in humans has not been determined.[20] The LD50 is 5.6 g/kg for rats and 20 g/kg for mice, indicating extremely low acute toxicity.[21] For comparison, in rats the LD50 of vitamin C is 12 g/kg and the LD50 of table salt is 3 g/kg.
Mechanisms of action
Piracetam’s mechanism of action, as with racetams in general, is not fully understood. The drug influences neuronal and vascular functions and influences cognitive function without acting as a sedative or stimulant.[5] Piracetam is a positive allosteric modulator of the AMPA receptor, although this action is very weak and its clinical effects may not necessarily be mediated by this action.[22] It is hypothesized to act on ion channels or ion carriers, thus leading to increased neuron excitability.[20]GABA brain metabolism and GABA receptors are not affected by piracetam[23]
Piracetam improves the function of the neurotransmitteracetylcholine via muscariniccholinergic (ACh) receptors[citation needed], which are implicated in memory processes.[24] Furthermore, piracetam may have an effect on NMDAglutamate receptors, which are involved with learning and memory processes. Piracetam is thought to increase cell membrane permeability.[24][25] Piracetam may exert its global effect on brain neurotransmission via modulation of ion channels (i.e., Na+, K+).[20] It has been found to increase oxygen consumption in the brain, apparently in connection to ATP metabolism, and increases the activity of adenylate kinase in rat brains.[26][27] Piracetam, while in the brain, appears to increase the synthesis of cytochrome b5,[28] which is a part of the electron transport mechanism in mitochondria. But in the brain, it also increases the permeability of some intermediates of the Krebs cycle through the mitochondrial outer membrane.[26]
Piracetam was first made some time between the 1950s and 1964 by Corneliu E. Giurgea.[31] There are reports of it being used for epilepsy in the 1950s.[32]
Society and culture
In 2009 piracetam was reportedly popular as a cognitive enhancement drug among students.[33]
Legal status
Piracetam is an uncontrolled substance in the United States meaning it is legal to possess without a license or prescription.[34]
Regulatory status
In the United States, piracetam is not approved by the Food and Drug Administration.[1] Piracetam is not permitted in compounded drugs or dietary supplements in the United States.[35] Nevertheless, it is available in a number of dietary supplements.[4]
In the United Kingdom, piracetam is approved as a prescription drug Prescription Only Medicine (POM) number is PL 20636/2524[36] for adult with myoclonus of cortical origin, irrespective of cause, and should be used in combination with other anti-myoclonic therapies.[37]
In Japan piracetam is approved as a prescription drug.[38]
Piracetam has no DIN in Canada, and thus cannot be sold but can be imported for personal use in Canada.[39]
In Hungary, piracetam was a prescription-only medication, but as of 2020, no prescription is required and piracetam is available as an over-the-counter drug under the name Memoril Mite, and is available in 600 mg pills.
According to the literature reports, the synthetic route of piracetam can be divided into four synthetic methods: α-pyrrolidone method, glycine method, succinic anhydride method and one-step synthesis method:[0009] I. α-pyrrolidone method, 2-pyrrolidone is a lactam, which can react with a strong base (sodium hydride or potassium hydride, sodium methoxide) to generate pyrrolidone metal salt, which can be further combined with halogenated ester or halogen Substitute amide reaction to generate N-alkylated product.[0010] In 1966, a method for preparing piracetam by reacting pyrrolidone and chloroacetamide in 1,4-dioxane with sodium hydrogen as a strong base was reported. The specific synthetic route is shown in Scheme 1:[0011]
[0012] In this process, due to the high price of dioxane, industrial production is still difficult. On the basis of the above process, Xu Yungen used dimethyl sulfoxide as the solvent and sodium methoxide as the acid binding agent to synthesize piracetam in the presence of the phase transfer catalyst benzyltriethylammonium chloride. Due to the difficulty of solvent recovery, the cost of this route is relatively high.[0013] In 1981, Zhou Renxing et al. used sodium methoxide as a strong base to extract methanol in toluene by fractional distillation to convert pyrrolidone into the corresponding sodium salt, and then react with ethyl chloroacetate. The resulting ethyl pyrrolidone ethyl acetate was subjected to ammonolysis. Piracetam can be produced. The specific synthetic route is shown in Scheme 2.[00141
[0015] Because the ammonolysis is carried out in a methanol solution of ammonia, the calculated amount of ethanol generated during the ammonolysis contaminates the methanol solution of ammonia used, which affects the recycling of the methanol solution of ammonia, and is therefore not conducive to process production.[0016] 2. Glycine method, glycine and its derivatives can be used as starting materials for the synthesis of pyroacetamide. Glycine can be prepared by γ-chlorination butylation, amination and cyclization.[0017] According to a British patent report in 1979, glycine trimethylsilyl ester was first condensed with γ-chlorobutyryl chloride, and the corresponding acid chloride was subjected to ammonolysis, and finally cyclized to produce piracetam. The specific synthesis method is as Scheme 3 Shown[0018]
[0019] In this type of synthesis route, some raw materials are not easily available, which restricts industrial production.[0020] 3. Succinic acid method, succinic acid is heated and dehydrated to generate succinic anhydride, succinic anhydride then reacts with glycine to generate an aminolysis product, and the aminolysis product is reduced by sodium tetrafluoroborate, and piracetam can be synthesized by aminolysis , The specific synthetic route is shown in SCheme4. [0021]
[0022] Because sodium tetrafluoroborate is used as a reducing agent, it is expensive, and it is difficult to expand the scale of industrial production. Succinimide generates sodium salt under the action of metal sodium, and its sodium salt reacts with chloroacetamide to generate N-alkylated product. The alkylated product can be electrolytically reduced to obtain piracetam. Since electrolytic reduction is still in the research stage in our country, the production cost of this method is relatively high.[0023] 4. One-step synthesis method, using ethyl 4-chloro-n-butyrate in the presence of sodium bicarbonate, using anhydrous ethanol as a solvent, and glycinamide hydrochloride under heating and refluxing to obtain piracetam in one step, The specific synthetic route is shown in S Cheme5.[0024]
[0025] In this route, glycinamide hydrochloride is very easy to absorb moisture and agglomerate to affect the reaction rate, and the reaction is not easy to control, so it is difficult to achieve industrial production.
With absolute ethanol as the solvent, ethyl 4-chloro-n-butanoate and glycinamide hydrochloride were refluxed for 20 h in the presence of sodium bicarbonate to obtain central stimulant piracetam. After recrystallization from isopropanol, the yield was about 58% with a purity of 99.6%.
Example 1[0036] A method for synthesizing piracetam, which includes the following steps:[0037] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionation column is connected with a thermometer, a condenser and a 500mL receiving flask. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 70°C, a methanol solution of sodium methoxide (28.4% (w/w); 114.0 g; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the dropwise addition is completed, the temperature is increased, and the normal pressure is distilled until the distillate is completely distilled out, and the reaction is completed.[0038] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. When the temperature of the reaction system drops to 60°C, a toluene solution of 58 mL (0.66 mol) of methyl chloroacetate is slowly added dropwise, and the reaction temperature is controlled to 80-100°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain α-pyrrolidone methyl acetate, and measure its content by HPLC (area normalization method). [C18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1 . OmL/min; detection wavelength is 205nm; injection volume is 20yL][0039] Preparation of Piracetam: Put about 130 mL of methanol in a 500 mL three-necked flask, and vent ammonia to saturation. The obtained ammonia/methanol solution was mixed with 100. Og α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 10 h, allowed to cool, filtered with suction, and the filter cake was dried.[0040] The purification of piracetam: 25.50g crude piracetam and 100mL isopropanol were sequentially added in a 500mL three-necked flask, heated to reflux for 40min, activated carbon was added, reflux stirring, hot filtration, and the resulting properties were all white As a powdery solid, the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 20.85 g of a white solid with a yield of 81.76% (calculated as α-pyrrolidone, the same below).Example 2[0042] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionation column is connected with a thermometer, a condenser and a 500mL receiving flask. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 100°C, a methanol solution of sodium methoxide (28.4% (w/w)); 114. Og; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the addition is complete, add toluene, increase the temperature, and distill at normal pressure until the distillate is completely distilled out, and the reaction is complete.[0043] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. When the temperature of the reaction system drops to 60°C, a mixed solution of 63 mL (0.72 mol) of methyl chloroacetate and 30 mL of toluene is slowly added dropwise, and the reaction temperature is controlled to 80-100°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain methyl α-pyrrolidone acetate, and measure its content by HPLC (area normalization method). [C18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1 .OmL/ min; detection wavelength is 205nm; injection volume is 20 μL][0044] Preparation of Piracetam: Put about 130 mL of methanol in a 250 mL three-necked flask, and ventilate ammonia to saturation. The obtained ammonia/methanol solution was mixed with 50.0 g of α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 12 hours, allowed to cool, filtered with suction, and the filter cake was dried.[0045] Purification of piracetam: 25.50g crude piracetam and 75mL methanol were sequentially added to a 500mL three-necked flask, heated to reflux for 40min, added activated carbon 0.5g, refluxed for 1h, hot filtered, magnetically stirred Under the conditions, the activated carbon was filtered out, and the properties were all white powdery solids, and the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 21.02g of white solids with a yield of 82.42%.Embodiment 3[0047] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionating column is connected with a thermometer, a condenser and a 1000 mL receiving bottle. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 70°C, a methanol solution of sodium methoxide (28.4% (w/w)); 114. Og; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the dropwise addition is completed, the temperature is increased, and the normal pressure is distilled until the distillate is completely distilled out, and the reaction is completed.[0048] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. A mixed solution of 79 mL (0.90 mol) of methyl chloroacetate and 50 mL of toluene was slowly added dropwise, and the reaction temperature was controlled to 70-90°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain methyl α-pyrrolidone acetate, and measure its content by HPLC (area normalization method). [C 18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1.0mL/min; The detection wavelength is 205nm; The injection volume is 20 μL)[0049] Preparation of Piracetam: Put about 130 mL of methanol in a 250 mL three-necked flask, and vent ammonia to saturation. The obtained ammonia/methanol solution was mixed with 100. Og α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 14h, allowed to cool, filtered with suction, and the filter cake was dried.[0050] Purification of piracetam: 25.50g crude piracetam and 125mL ethanol were sequentially added in a 500mL three-necked flask, heated to reflux for 40min, added activated carbon 0.5g, refluxed for 1h, hot filtered, magnetically stirred Activated carbon was filtered off under conditions to obtain white powdery solids in all properties, and the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 20.24 g of white solids with a yield of 79.37%.Example 4[0052] Preparation of α-pyrrolidone sodium salt: A 1000 mL three-necked flask was equipped with mechanical stirring, a constant pressure dropping funnel and a thorn-shaped fractionating column. The upper end of the fractionation column is connected with a thermometer, a condenser and a 500mL receiving flask. Under mechanical stirring, 46 mL (0.60 mol) of α-pyrrolidone and 250 mL of toluene were sequentially added to the three-necked flask. When the temperature of the reaction system reached 60°C, a methanol solution of sodium methoxide (28.4% (w/w); 114.0 g; 0.60 mol) was added dropwise under reduced pressure, and the distillate was collected. After the dropwise addition is completed, the temperature is increased, and the normal pressure is distilled until the distillate is completely distilled out, and the reaction is completed.[0053] Preparation of α-pyrrolidone methyl acetate: remove the fractionation device, connect a thermometer and a condenser, and connect a dropping funnel above the condenser. A mixed solution of 105 mL (1.20 mol) of methyl chloroacetate and 70 mL of toluene was slowly added dropwise, and the reaction temperature was controlled to be 60~70°C. Oh。 After the addition is complete, the insulation reaction is 5. Oh. Cool to room temperature, filter with suction, and distill the filtrate under reduced pressure. Collect the fraction (18mmHg) at 100~105°C to obtain methyl α-pyrrolidone acetate, and measure its content by HPLC (area normalization method). [C 18 column (4.6mmX 200mm, 5 μm) was used for purity determination; acetonitrile-dipotassium hydrogen phosphate/phosphate buffer solution (10:90) was used as the mobile phase (the pH value of phosphoric acid was adjusted to 6.0); the flow rate was 1.0mL/min; The detection wavelength is 205nm; The injection volume is 20 μL)[0054] Preparation of Piracetam: Put about 130 mL of methanol in a 500 mL three-necked flask, and ventilate ammonia to saturation. The obtained ammonia/methanol solution was mixed with 100. Og α-pyrrolidone methyl acetate and placed in a reaction kettle, reacted at 50~65°C for 16h, allowed to cool, filtered with suction, and the filter cake was dried.[0055] The purification of piracetam: 25.50g crude piracetam and 100mL methanol were sequentially added into a 500mL three-necked flask, heated to reflux for 40min, added activated carbon, refluxed for dissolution, hot filtered, and the properties were all white powders The solid, the filter cake was dried overnight at 50°C in a vacuum drying oven to obtain 20.69 g of a white solid, with a yield of 81. 13%.[0056] Chemical analysis of the white crystals synthesized in each of the foregoing examples, and the obtained physical property values are as follows, thereby confirming that the synthesized product is piracetam.[0057] Melting point: 151.6-152. (TC[0058] ESI-MS m / z: 165. 06 [M + Na] +[0059] 1H-NMR (400MHz, DMS〇-d6, ppm) δ : 7. 38 (s, 1H), 7. 09 (s, 1H), 3. 74 (s, 2H), 3. 36 (t, J =7. 08Hz, 2H), 2. 23 (t, J = 7. 84Hz, 2H), I. 93 (m, 2H).[0060] 13C-NMR(100MHz, DMS0-d6, ppm) δ : 17. 80, 30. 42, 45. 28, 47. 74, 170. 21,174. 90. PATENTCN110903230A *2019-12-042020-03-24Beijing Yuekang Kechuang Pharmaceutical Technology Co., Ltd.An industrialized preparation method of Pramiracetam sulfate PATENTCN104478779A2015-04-01New synthetic method of nootropic drug Piracetam
^ Farooq MU, Min J, Goshgarian C, Gorelick PB (September 2017). “Pharmacotherapy for Vascular Cognitive Impairment”. CNS Drugs(Review). 31 (9): 759–776. doi:10.1007/s40263-017-0459-3. PMID28786085. S2CID23271739. Other medications have been considered or tried for the treatment of VCI or VaD. These include […] piracetam. There is no convincing evidence about the efficacy of these medications in the treatment of VCI.
^ Zhang J, Wei R, Chen Z, Luo B (July 2016). “Piracetam for Aphasia in Post-stroke Patients: A Systematic Review and Meta-analysis of Randomized Controlled Trials”. CNS Drugs. 30 (7): 575–87. doi:10.1007/s40263-016-0348-1. PMID27236454. S2CID22955205.
^ Yeo SH, Lim ZI, Mao J, Yau WP (October 2017). “Effects of Central Nervous System Drugs on Recovery After Stroke: A Systematic Review and Meta-Analysis of Randomized Controlled Trials”. Clinical Drug Investigation. 37 (10): 901–928. doi:10.1007/s40261-017-0558-4. PMID28756557. S2CID6520934.
^ Chouinard G, Annable L, Ross-Chouinard A, Olivier M, Fontaine F (1983). “Piracetam in elderly psychiatric patients with mild diffuse cerebral impairment”. Psychopharmacology. 81 (2): 100–106. doi:10.1007/BF00429000. PMID6415738. S2CID32702769.
^ Hakkarainen H, Hakamies L (1978). “Piracetam in the treatment of post-concussional syndrome. A double-blind study”. European Neurology. 17 (1): 50–55. doi:10.1159/000114922. PMID342247.
^ Jump up to:ab Winnicka K, Tomasiak M, Bielawska A (2005). “Piracetam–an old drug with novel properties?”. Acta Poloniae Pharmaceutica. 62(5): 405–9. PMID16459490.
^ Müller WE, Eckert GP, Eckert A (March 1999). “Piracetam: novelty in a unique mode of action”. Pharmacopsychiatry. 32 (Suppl 1): 2–9. doi:10.1055/s-2007-979230. PMID10338102.
^ Jump up to:ab Grau M, Montero JL, Balasch J (1987). “Effect of Piracetam on electrocorticogram and local cerebral glucose utilization in the rat”. General Pharmacology. 18 (2): 205–11. doi:10.1016/0306-3623(87)90252-7. PMID3569848.
^ Nickolson VJ, Wolthuis OL (October 1976). “Effect of the acquisition-enhancing drug piracetam on rat cerebral energy metabolism. Comparison with naftidrofuryl and methamphetamine”. Biochemical Pharmacology. 25 (20): 2241–4. doi:10.1016/0006-2952(76)90004-6. PMID985556.
^ Tacconi MT, Wurtman RJ (1986). “Piracetam: physiological disposition and mechanism of action”. Advances in Neurology. 43: 675–85. PMID3946121.
^ Yeh HH, Yang YH, Ko JY, Chen SH (July 2006). “Rapid determination of piracetam in human plasma and cerebrospinal fluid by micellar electrokinetic chromatography with sample direct injection”. J Chromatogr A. 1120 (1–2): 27–34. doi:10.1016/j.chroma.2005.11.071. PMID16343512.
^ Bravo-Martínez J, Arenas I, Vivas O, Rebolledo-Antúnez S, Vázquez-García M, Larrazolo A, García DE (October 2012). “A novel CaV2.2 channel inhibition by piracetam in peripheral and central neurons”. Exp Biol Med (Maywood). 237 (10): 1209–18. doi:10.1258/ebm.2012.012128. PMID23045722.
Piroxicam is a nonsteroidal anti-inflammatory drug (NSAID) of the oxicam class used to relieve the symptoms of painful inflammatory conditions like arthritis.[3][4] Piroxicam works by preventing the production of endogenous prostaglandins] which are involved in the mediation of pain, stiffness, tenderness and swelling.[3] The medicine is available as capsules, tablets and (not in all countries) as a prescription-free gel 0.5%.[5] It is also available in a betadex formulation, which allows a more rapid absorption of piroxicam from the digestive tract.[3] Piroxicam is one of the few NSAIDs that can be given parenteral routes.
It was patented in 1968 by Pfizer and approved for medical use in 1979.[6] It became generic in 1992,[7] and is marketed worldwide under many brandnames.[1]
Medical uses
It is used in the treatment of certain inflammatory conditions like rheumatoid and osteoarthritis, primary dysmenorrhoea, postoperative pain; and act as an analgesic, especially where there is an inflammatory component.[3] The European Medicines Agency issued a review of its use in 2007 and recommended that its use be limited to the treatment of chronic inflammatory conditions, as it is only in these circumstances that its risk-benefit ratio proves to be favourable.[5][8]
In October 2020, the U.S. Food and Drug Administration (FDA) required the drug label to be updated for all nonsteroidal anti-inflammatory medications to describe the risk of kidney problems in unborn babies that result in low amniotic fluid.[9][10] They recommend avoiding NSAIDs in pregnant women at 20 weeks or later in pregnancy.[9][10]
The project that produced piroxicam began in 1962 at Pfizer; the first clinical trial results were reported in 1977, and the product launched in 1980 under the brand name “Feldene”.[7][12] Major patents expired in 1992[7] and the drug is marketed worldwide under many brandnames.[1]
https://patents.google.com/patent/CN101210013A/enIn the glassed steel reaction vessels of 2000L, add first ethyl ester thing 140Kg, dimethylbenzene 1500L, silica gel 10Kg.Be warming up to 100 ℃ of amino pyrrole 52Kg of adding 2-, continue to be warming up to the solvent refluxing temperature, keep refluxing slowly, steam the ethanol of reaction generation and the mixture of dimethylbenzene simultaneously, TLC follows the tracks of reaction, and reaction in 4.5-5 hour finishes.Underpressure distillation, the control temperature in the kettle is no more than 70 ℃, when the system volume be about cumulative volume 1/3 the time stop distillation, be cooled to normal temperature, stir 6-8h and filter, be i.e. crude product.Crude product adds methyl alcohol 1500L and adds the 15Kg gac, refluxes 30 minutes, filters, and is cooled to normal temperature, stirs 6-8h, methyl alcohol drip washing, 60-70 ℃ is dried by the fire 3-5h, measure product 140.5Kg, yield 85%.Press Cp2005 version standard detection, outward appearance; Off-white color, content 〉=99%.Methanol mother liquor reclaims methyl alcohol to overall 1/3 o’clock, and cooling stirring at normal temperature 6-8h filters and collects product, oven dry measure product 10Kg, yield 5.7%, this product meet the Cp2005 version and require to add up to yield.Add up to yield 90.7%.PAPER Bulletin of the Korean Chemical Society, 26(11), 1771-1775; 2005
^ Jump up to:abcdefg Brayfield, A, ed. (14 January 2014). “Piroxicam”. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 24 June 2014.
^ Jump up to:abc Lombardino, JG; Lowe, JA 3rd (2004). “The role of the medicinal chemist in drug discovery–then and now”. Nat Rev Drug Discov. 3 (10): 853–62. doi:10.1038/nrd1523. PMID15459676. S2CID11225541.. See: [1] Box 1: Discovery of piroxicam (1962–1980)
AUR-101, a ROR gamma inverse agonist for autoimmune disorders like psoriasis
AUR-101 is an ROR-gammaT inverse agonist in phase II clinical development at Aurigene for the treatment of patients with moderate-to-severe chronic plaque-type psoriasis.
Aurigene Announces First Patient Dosed with AUR101 in Phase II Study in Patients with Moderate to Severe Psoriasis
Bangalore, February 17, 2020 — Aurigene, a development stage biotechnology company, today announced dose administration for the first patient in INDUS-2, a Phase II double blind placebo-controlled three-arm study of AUR101 in patients with moderate to severe psoriasis. AUR101 is an oral small molecule inverse agonist of RORγ and has shown desirable pharmacodynamic modulation of IL-17 and acceptable safety in a completed Phase I human study conducted in Australia.
“The initiation of this Phase II study under a US FDA IND represents a significant milestone for Aurigene, as it marks the first program which Aurigene has led from the bench side to the clinic all by itself,” said Murali Ramachandra, PhD, Chief Executive Officer of Aurigene. “We look forward to producing important clinical data by the end of 2020 to guide our future development plans and demonstrating Aurigene’s unique expertise in conducting Proof-of-Concept studies in a quality and fast-paced manner.”
About AUR101-201 and the Phase II Study of AUR101 in Patients with Moderate to Severe Psoriasis
The purpose of the Phase II multi-center, blinded, placebo-controlled, three-arm study is to evaluate the clinical activity of AUR101 in patients with moderate to severe psoriasis. In two of the arms, AUR101 will be administered twice daily, at 400 mg PO BID and 600 mg PO BID, for 12 weeks. Patients in the third arm will receive matched blinded placebo in a double dummy fashion. The trial is listed at clinicaltrials.gov with identifier NCT04207801.
About Aurigene
Aurigene is a development stage biotech company engaged in discovery and clinical development of novel and best-in-class therapies to treat cancer and inflammatory diseases and a wholly owned subsidiary of Dr. Reddy’s Laboratories Ltd. (BSE: 500124, NSE: DRREDDY,NYSE: RDY). Aurigene is focused on precision- oncology, oral immune checkpoint inhibitors, and the Th-17 pathway. Aurigene currently has several programs from its pipeline in clinical development. Aurigene has also submitted an IND to DCGI, India for a Phase IIb/III trial of CA-170, a dual inhibitor of PD-L1 and VISTA, in non-squamous NSCLC. Additionally, Aurigene has multiple compounds at different stages of pre-clinical development. Aurigene has partnered with many large and mid-pharma companies in the United States and Europe and has 15 programs currently in clinical development. For more information, please visit Aurigene’s website at https://www.aurigene.com/.
CLIP
Signalling of multiple interleukin (IL)-17 family cytokines via IL-17 receptor A drives psoriasis-related inflammatory pathways
M.A.X. Tollenaere,J. Hebsgaard,D.A. Ewald,P. Lovato,S. Garcet,X. Li,S.D. Pilger,M.L. Tiirikainen,M. Bertelsen,J.G. Krueger,H. Norsgaard,First published: 01 April 2021 https://doi.org/10.1111/bjd.20090Citations: 2Funding sources LEO Pharma A/S funded this study.Conflicts of interest M.A.X.T., J.H., D.A.E., P.L., S.D.P., M.L.T., M.B. and H.N. are employees of LEO Pharma. J.G.K. received grants paid to his institution from Novartis, Pfizer, Amgen, Lilly, Boehringer, Innovaderm, BMS, Janssen, AbbVie, Paraxel, LEO Pharma, Vitae, Akros, Regeneron, Allergan, Novan, Biogen MA, Sienna, UCB, Celgene, Botanix, Incyte, Avillion and Exicure; and personal fees from Novartis, Pfizer, Amgen, Lilly, Boehringer, Biogen Idec, AbbVie, LEO Pharma, Escalier, Valeant, Aurigene, Allergan, Asana, UCB, Sienna, Celgene, Nimbus, Menlo, Aristea, Sanofi, Sun Pharma, Almirall, Arena and BMS.Data Availability Statement The gene array dataset described in this publication has been deposited in NCBI’s Gene Expression Omnibus and is accessible through GEO Series accession number GSE158448 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE158448).
Although biologics such as anti-TNFα antibody are fairly successful in the treatment of autoimmune disorders, there is significant unmet need due to heterogeneity in diseases and lack of response to established therapies in some patients. While biologics typically target one cytokine signaling pathway, small molecule therapeutics directed towards intracellular target(s) can interfere in the signaling from multiple cytokines potentially leading to improved response. Development of small molecule oral inhibitors of IRAK4 and RORgamma to target TLR/IL-R and Th17 pathway respectively will be discussed.
This application claims the benefit of Indian provisional application number 5641/CHE/2013 filed on 06th December 2013 which hereby incorporated by reference.
(5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid (5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydrofuran-2-yl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid 106560-14-9[RN] 4-Thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid, 6-[(1R)-1-hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-, (5R,6S)- 6α-[(R)-1-hydroxyethyl]-2-[(R)-tetrahydrofuran-2-yl]pen-2-em-3-carboxylic acid 4-Oxofenretinide 4-Oxo-N-(4-hydroxyphenyl)retinamide 6α-[(1R)-1-hydroxyethyl]-2-[(2R)-tetrahydrofuran-2-yl]-2,3-didehydropenam-3-carboxylic acid 7305146 [Beilstein] FaropenemCAS Registry Number: 106560-14-9 CAS Name: (5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid Additional Names: fropenem; (5R,6S,8R,2¢R)-2-(2¢-tetrahydrofuryl)-6-hydroxyethylpenem-3-carboxylate Molecular Formula: C12H15NO5S Molecular Weight: 285.32 Percent Composition: C 50.51%, H 5.30%, N 4.91%, O 28.04%, S 11.24% Literature References: Orally active, b-lactamase stable, penem antibiotic.Prepn: M. Ishiguro et al.,EP199446; eidem,US4997829 (1986, 1991 both to Suntory); eidem,J. Antibiot.41, 1685 (1988).Pharmacokinetics: A. Tsuji et al.,Drug Metab. Dispos.18, 245 (1990). In vitro antimicrobial spectrum: J. M. Woodcock et al.,J. Antimicrob. Chemother.39, 35 (1997). b-Lactamase stability: A. Dalhoff et al., Chemotherapy (Basel)49, 229 (2003).HPLC determn in plasma: R. V. S. Nirogi et al., Arzneim.-Forsch.55, 762 (2005). Clinical trial in urinary tract infections: S. Arakawa et al.,Nishinihon J. Urol.56, 300 (1994); in bacterial sinusitis: R. Siegert et al., Eur. Arch. Otorhinolaryngol.260, 186 (2003). Derivative Type: Sodium salt CAS Registry Number: 122547-49-3 Additional Names: Furopenem Manufacturers’ Codes: ALP-201; SUN-5555; SY-5555; WY-49605 Trademarks: Farom (Daiichi) Molecular Formula: C12H15NNaO5S Molecular Weight: 308.31 Percent Composition: C 46.75%, H 4.90%, N 4.54%, Na 7.46%, O 25.95%, S 10.40% Properties: [a]D22 +60° (c = 0.10). Optical Rotation: [a]D22 +60° (c = 0.10) Derivative Type: Daloxate CAS Registry Number: 141702-36-5 CAS Name: (5R,6S)-6-[(1R)-1-Hydroxyethyl]-7-oxo-3-[(2R)-tetrahydro-2-furanyl]-4-thia-1-azabicyclo[3.2.0]hept-2-ene-2-carboxylic acid (5-methyl-2-oxo-1,3-dioxol-4-yl)methyl ester Additional Names: faropenem medoxomil Manufacturers’ Codes: Bay-56-6854; SUN-208 Trademarks: Orapem (Replidyne) Molecular Formula: C17H19NO8S Molecular Weight: 397.40 Percent Composition: C 51.38%, H 4.82%, N 3.52%, O 32.21%, S 8.07% Literature References: Prepn: H. Iwata et al., WO9203442; eidem, US5830889 (1992, 1998 both to Suntory). Properties: Pale yellow crystals. Therap-Cat: Antibacterial (antibiotics). Keywords: Antibacterial (Antibiotics); ?Lactams; Penems.
The sodium salt faropenem sodium, available under the trade name Farom, has been marketed in Japan since 1997. (CID 636379 from PubChem)
The prodrug form faropenem medoxomil[4] (also known as faropenem daloxate) has been licensed from Daiichi Asubio Pharma by Replidyne, which plans to market it in conjunction with Forest Pharmaceuticals. The trade name proposed for the product was Orapem, but company officials recently announced this name was rejected by the FDA.[5]
Clinical use
As of 8 September 2015, Faropenem has yet to receive marketing approval in the United States, and was submitted for consideration by the United States Food and Drug Administration (FDA) on 20 December 2005. The new drug application dossier submitted included these proposed indications:
acute bacterial sinusitis
community-acquired pneumonia
acute exacerbations of chronic bronchitis
uncomplicated skin and skin structure infections
urinary tract infections
History
The FDA refused to approve faropenem, an antibiotic manufactured by Louisville-based Replidyne. The FDA said the drug was “nonapprovable”, but did not refer to specific safety concerns about the product. The company will have to conduct new studies and clinical trials, lasting an estimated two more years, to prove the drug treats community-acquired pneumonia, bacterial sinusitis, chronic bronchitis, and skin infections.[citation needed]
In India it is available as Farobact 200/300ER CIPLA.
PATENT
https://patents.google.com/patent/WO2008035153A2/enFaropenem is an orally active β-lactam antibiotic belonging to the penem group. Faropenem is chemically known as 6-(l-hydroxyethyl)-7-oxo-3-(oxolan-2-yl)-4-thia-l-azabicyclo[3.2.0]hept-2-ene-2-carboxylicacid. The known forms of Faropenem are Faropenem sodium and the prodrug form, FaropenemMedoxomil (also known as Faropenem Daloxate). In view of the importance of the compound of the formula (I), several synthetic procedures to prepare the compound have been reported.US 4,997,829 provides process for the preparation of faropenem according to the following scheme. The process is exemplified with the allyl protected carboxyl group. One of the process involves the reaction of A- acetoxyazetidinone with tetrahydrothiofuroic acid, condensation with allyl glyoxalate in refluxing benzene, chlorination with thionyl chloride, reaction of triphenylphosphine with lutidine in hot THF, cyclization in refluxing toluene, deprotection of silyl protecting group with tetrabutylammonium fluoride, treating with triphenylphosphine and, treating with sodium 2-ethylhexanoate and (PP^)4Pd to result faropenem sodium. The process exemplified utilizes benzene as solvent, which is not environmentally acceptable. Tetrabutylammonium fluoride was used as desilylating agent that is expensive. Even though the description teaches that optically active compounds can be employed, the examples utilized the dl-compound of tetrahydrothiofuroic acid further requiring resolution.
Methods are provided for the synthesis of series of penem compounds in J Antibiotics 1988, 41(11), 1685-1693. The provided methods utilize sulfonylazetidinone as the starting materials. As one of the procedures gives lesser yield, another procedure was adopted which uses silver salts.Japanese patent, JP2949363 describes a process for deallylation and salt formation with an alkali metal salt of carboxylic acid in the presence of a catalytic amount of palladium complex for the preparation of faropenem.EP410727 describes a process for removing allyl group from a penem compound using cyclic 1,3-diketone such as dimedone.The yield and quality of the final product is always less in the above prior art methods. With the continued research, the present inventors have undertaken extensive studies for developing a process for the preparation of compound of formula (I), which is commercially viable, involves simple techniques such as crystallizations, with improved yields and quality of the product, and with lesser reaction time. None of the prior art suggests or teaches the techniques provided herein.The process is shown in Scheme-I as given below:
One-pot process for the preparation of Faropenem sodium:Sodium salt of R(+)-tetrahydrofuran-2-thiocarboxylic acid (67 g) in aqueous acetone was added slowly to a solution of AOSA (100 g) in acetone (200 mL) and stirred for 3 h at pH 8.0 to 8.5 using sodium bicarbonate solution.After completion of the reaction, the product was extracted with toluene. The combined toluene layer was washed with saturated sodium bicarbonate solution and brine solution. Toluene was removed under vacuum completely and the mass obtained, 3-(l’-tert-butyldimethylsilyloxyethyl)-4-(2′- tetrahydrofuranoylthio)-2-azetidinone was directly taken for next step.3-(r-tert-Butyldimethylsilyloxyethyl)-4-(2′-tetrahydrofuranoylthio)-2- azetidinone obtained was dissolved in toluene (1000 mL) and cooled to -10 to -5 °C under nitrogen. Triethylamine (124 mL) was added to it followed by allyl oxalyl chloride (82 g) at -10 to- 5 0C for 2 h. After completion of the reaction, cold water was added to the mass and washed with dilute hydrochloric acid and sodium bicarbonate solution. Toluene layer was separated and washed with purified water. The toluene layer containing compound of formula (VI) was concentrated under vacuum at 50 to 60 °C and taken for next step as such.Compound of formula (VI) (150 g) was dissolved in triethyl phosphite (150 mL), heated to 60 0C and stirred under nitrogen atmosphere. Toluene (3000 mL) was added, heated to 100 to 110 °C and stirred for 20- 24 h. Toluene was distilled under vacuum completely. Product obtained, allyl (1 ‘R,2″R,5R,6S)-6-(l 5-tert-butyldimethylsilyloxyethyl)-2-(2″-tetrahydrofuranyl) penem-3-carboxylate (VII) was directly taken for next step.Compound (VII) obtained was dissolved in DMF (700 mL) at 30 °C.Ammonium hydrogen difluoride (80 g) and NMP (210 mL) were added and stirred at room temperature for 25 to 35 h. The reaction mass was quenched into a mixture of water-ethyl acetate and stirred at room temperature. The ethyl acetate layer was separated and the aqueous layer extracted with ethyl acetate. ■ The combined ethyl acetate layer was washed with water followed by saturated sodium bicarbonate solution. The ethyl acetate layer was charcoal treated. The ethyl acetate layer containing allyl (l’R,2″R,5R,6S)-6-(l’-hydroxyethyl)-2-(2″- tetrahydrofuranyl)penem-3-carboxylate (XII) was partially distilled and taken for the next step.The ethyl acetate layer containing compound of formula (XII), Pd/C, sodium bicarbonate and purified water (1000 mL) were taken in an autoclave and maintained 5 to 10 kg pressure of hydrogen gas for 2-5 h. After completion of the reaction the Pd/C was filtered off and ethyl acetate layer separated. The pH of the mass was adjusted to 1.5 and extracted with ethyl acetate. The aqueous layer was extracted again with ethyl acetate twice. The combined ethyl acetate layer was carbon treated. Sodium-2-ethylhexanoate in ethyl acetate was added slowly and stirred. The precipitated title compound was filtered under vacuum, washed with acetone and dried. Dry weight of the product: 65-75 g.Example 9Purification of Faropenem sodiumCrude Faropenem sodium (50 g) was dissolved in purified water (200 mL) at 25-30 0C. The solution was charcoalised. Acetone (1500 mL) was added. The reaction mass was stirred further for 10 min. The precipitated solid was cooled to 0 —2 °C then filtered, washed with acetone and dried at room temperature. Weight of pure Faropenem sodium is 43 to 46 g (Purity 99.95%).Example 9aPurification of Faropenem sodiumCrude Faropenem sodium (50 g) was dissolved in purified water (200 mL) at 25-30 °C. Acetone (150O mL) was added. The reaction mass was stirred further for 10 min. The precipitated solid was cooled to 0-2 °C then filtered, washed with acetone and dried at room temperature. Weight of pure Faropenem sodium is 43 to 46 g (Purity 99.95%).
PATENT
https://patents.google.com/patent/CN103880864B/enFaropenem sodium is developed by Japanese Suntory companies, and first penemss antibiosis in listing in 1997 Element, it are similar to the several carbapenem antibiotics for listing, strong with has a broad antifungal spectrum, antibacterial activity, to beta-lactamase Stably, the features such as also having good action to extended spectrumβ-lactamase producing strains, citrobacter, enterococcus and anaerobe etc.. It is first orally active, penems antibiotics stable to beta-lactamase in the world so far.Its structural formula As follows: Report about Faropenem sodium preparation method is a lot, mainly has several as follows:1st, J. Antibiotics 1988, the method that reports in 41,1685, see below row reaction equation: Acyl group substitution reaction is carried out in the basic conditions with 4-AA and three beneze methane thiols and obtains thio trityl as protecting group Aza cyclo-butanone, then when 2-TETRAHYDROFUROYL chlorine is connected with lactams, using silver nitrate as condensing agent, but nitric acid Silver is expensive, and cost is too high, while the silver chloride for generating is difficult to filter, is not suitable for large-scale production.2nd, the classical preparation method of United States Patent (USP) US4997829 report:There is acyl with (R) tetrahydrofuran -2- thiocarboxylic acids Base substitution reaction generates thioesters, then through condensation, chlorine replacement, intramolecular Witting cyclization, slough hydroxyl protecting group and carboxylic Base protection group obtains product, and this synthetic route yield is very low, while side chain is thio-compoundss, abnormal smells from the patient is extremely smelly, and prepares complexity, There is-fixed harm to human body and environment.It is also required in chloro building-up process using pungent thionyl chloride, these factors are all It is unfavorable for industrialized production 3rd, the method that reports in Chinese patent CN1314691 is as follows: Said method route is shorter, is produced using one kettle way, more convenient.But said method is related to some other salt such as acetate using heavy metal palladium in last operation The deprotecting regent of compound and triphenyl phosphorus together as pi-allyl, metal palladium reagent is expensive, while triphenyl phosphorus are most More difficult removing in step afterwards, increases operation difficulty, affects product quality.Allyloxy is used easily to produce as protection group simultaneously A kind of double bond olefinic polymerization species impurity of life, affects product quality, reduces yield.Embodiment one(R) tetrahydrofuran -2- thiocarboxylic acids (198g, 1.5 mol) are put in 3L reaction bulbs, plus 1 mol/L hydrogen-oxygens Change sodium body lotion (I.5 L) to be adjusted at 5 DEG C of pH 9- 10,0-, Deca 4AA(287g, 1. 0mo l) acetone (1 L) Solution, drop are finished, and are adjusted to pH 8 or so, 2 h of room temperature reaction with 1 mol/L sodium hydroxide. and add water (500 ml) dilution, second Acetoacetic ester (600 ml x3) is extracted, and merges organic layer, successively with 5 % sodium bicarbonate solutions (300 ml x 2) and water (300 m1 x 2) is washed, and anhydrous sodium sulphate is dried, and is filtered, and filtrate concentrates, and obtains pale yellow oil (about 360 g), directly Input the next step.Embodiment twoThe mixing of concentrated solution as obtained above, triethylamine (l70g, 1.7 mol) and dichloromethane (1.5 L), 0-5 DEG C Deca chlorine oxalic acid is finished to p-Nitrobenzyl (414.1 g, 1 .7 mo l), drop, and equality of temperature reacts 2 h, and add water (1 L) dilution, Extracted with dichloromethane (500 ml x 4), merge organic layer, molten with water (300m1 x 2) and 5 % sodium bicarbonate successively Liquid (300 m1 x 2) is washed, anhydrous sodium sulfate drying, is filtered, and concentration obtains pale yellow oil (about 530g), direct plunges into The next step..Embodiment threeAbove-mentioned gained grease, dimethylbenzene (4L) and NSC 5284 (500ml) are mixed, heating reflux reaction 5h , reduce pressure and boil off dimethylbenzene and NSC 5284, residue ethyl acetate-hexane (1:5,1 L) recrystallization, obtain yellowish Color solid (334.3g, 61%, in terms of 4AA).Example IVAbove-mentioned solid (0.60 mol of 330g.) is dissolved in methanol (2 L), adds 1.0M hydrochloric acid (0.4 L), adds palladium carbon (15.0 g), hydrogen is passed through, 40 DEG C of stirrings, response time are 16 h, and the pressure of system is 4atm, after reaction terminates, crosses and filters Catalyst is removed, is concentrated.Embodiment fiveThe product obtained after above-mentioned concentration is dissolved in tetrahydrofuran 600ml, the 2 ethyl hexanoic acid sodium of 100.0g is added Tetrahydrofuran(200ml)And water(200 ml)Mixed solution, 2 h are stirred at room temperature, have faint yellow solid generate, filter, be method Faropenem crude product 147.0g.Embodiment sixBy above-mentioned solid deionized water(2200ml)Acetone is slowly added under dissolved solution, stirring to start to become to solution Muddiness, when about adding acetone 750ml, solution starts to become cloudy, and stops adding, and continues stirring and allows its crystallize overnight, sucking filtration, acetone Washing, dries, and obtains the Faropenem sodium fine work 125.0g of white.
Syn
AU 8654460; EP 0199446; JP 1994128267; US 4997829
This compound is prepared by several related ways: 1) The reaction of silylated azetidinone (I) with tetrahydrofuran-2-thiocarboxylic acid (II) by means of NaOH in THF – water gives the azetidinone thioester (III), which is condensed with allyl glyoxylate in refluxing benzene yielding the hydroxyester (IV). The reaction of (IV) with SOCl2 affords the chloroester (V), which by reaction with triphenylphosphine by means of lutidine in hot THF is converted into the phosphoranylidene derivative (VI). The elimination of the silyl protecting group of (VI) with tetrabutylammonium fluoride gives the azetidinone (VII), which is cyclized in refluxing toluene yielding the (5R,6S)-6-[1(R)-hydroxyethyl]-2-[2(R)-tetrahydrofuryl]penem-3-carboxyli c acid allyl ester (VIII). Finally, this compound is hydrolyzed with triphenylphosphine, sodium 2-ethylhexanoate and Pd-tetrakis(triphenylphosphine). 2) The condensation of the silver salt of protected azetidinone (IX) with tetrahydrofuran-2(R)-carbonyl chloride (X) also yields the phosphoranylidene salt (VI). 3) Phosphoranylidene ester (VI) can also be cyclized first in refluxing benzene yielding the silylated penem ester (XI), which is deprotected with tetrabutylammonium fluoride to (VIII). 4) The hydrolysis of allyl ester (VIII) to the final product can also be performed with paladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)-5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethylacetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene, or ethylene glycol dimethyl ether. 5) The preceding hydrolysis can also be performed with triphenylphosphine and paladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone.
Treatment of the silylated azetidinone (I) with tritylmercaptan affords the tritylsulfanyl-azetidinone (II), which is converted into the silver salt (III) by reaction with AgNO3. Compound (III) is coupled with tetrahydrofuran-2(R)-carbonyl chloride (IV) — obtained by treatment of carboxylic acid (V) with thionyl chloride — providing the azetidinone thioester (VI). Coupling of azetidinone (VI) with allyl oxalyl chloride (VII) in CH2Cl2 by means of Et3N, followed by intramolecular Wittig cyclization by means of triethyl phosphite in refluxing xylene, affords penem (VIII). Alternatively, compound (VIII) can also be obtained as follows: Substitution of phenyl sulfonyl group of azetidinone (X) by tritylmercaptan by means of NaOH in acetone/water provides tritylsulfanyl-azetidinone (XI), which is condensed with allyl oxalyl chloride (VII) by means of DIEA in CH2Cl2 to give the oxalyl amide (XII). Compound (XII) is then treated with AgNO3 and pyridine in acetonitrile, providing the silver mercaptide (XIII), which is acylated with tetrahydrofuran-2(R)-carbonyl chloride (IV) in acetonitrile to afford the penem precursor (XIV). Penem (VIII) is obtained by intramolecular Wittig cyclization of (XIV) with P(OEt)3 in refluxing xylene. Finally, faropenem sodium can be obtained by removal of the tbdms protecting group of (VIII) by means of either Et3N tris(hydrogen fluoride) in ethyl acetate or tetrabutylammonium fluoride (TBAF) and HOAc in THF to give compound (IX). This is followed by allyl ester group removal of (IX), which can be performed under several different conditions: i) triphenylphosphine, sodium 2-ethylhexanoate and palladium tetrakis(triphenylphosphine); ii) palladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)-5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethyl acetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene or ethylene glycol dimethyl ether; iii) triphenylphosphine and palladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone; or iv) palladium acetate in the presence of P(OBu)3 and sodium propionate in THF.
Treatment of the silylated azetidinone (I) with tritylmercaptan affords the tritylsulfanylazetidinone (II), which by reaction with AgNO3 is converted into the silver salt (III). Compound (III) is coupled with tetrahydrofuran-2(R)-carbonyl chloride (IV) ?obtained by treatment of carboxylic acid (V) with thionyl chloride ?to provide the azetidinone thioester (VI). Alternatively, compound (VI) can be obtained by condensation of tetrahydrofuran-2(R)-thiocarboxylic S-acid (VII) ?obtained by treatment of carboxylic acid (V) with hydrogen sulfide ?with silylated azetidinones (I) or (VIII) by means of NaOH in THF/water. Condensation of azetidinone thioester (VI) with allyl glyoxylate (IX) in refluxing benzene gives the hydroxy ester (X), which is treated with SOCl2 to yield the chloro ester (XI). Reaction of compound (XI) with triphenylphosphine and lutidine in hot THF provides the phosphoranylidene derivative (XII), which is converted into (5R,6S)-6-[1(R)-hydroxyethyl]-2-[2(R)-tetrahydrofuryl]penem-3-carboxylic acid allyl ester, faropenem allyl ester (XIII) by removal of the silyl protecting group with tetrabutylammonium fluoride, followed by cyclization in refluxing toluene. Compound (XII) can also be obtained by condensation of the silver salt of protected azetidinone (XIV) with tetrahydrofuran-2(R)-carbonyl chloride (V).
Alternatively, faropenem allyl ester (XIII) can also be prepared by cyclization of compound (XII) in refluxing benzene to yield silylated penem allyl ester (XV), which is then deprotected with either tetrabutylammonium fluoride in AcOH or triethylamine tris(hydrogen fluoride) in methyl isobutyl ketone or toluene. Penem (XV) can also be synthesized by several related ways: a) By coupling of azetidinone (VI) with allyl oxalyl chloride (XVI) in CH2Cl2 by means of Et3N, followed by intramolecular Wittig cyclization by means of triethyl phosphite in refluxing xylene. b) Substitution of phenyl sulfonyl group of azetidinone (VIII) by tritylmercaptan by means of NaOH in acetone/water provides tritylsulfanyl-azetidinone (II), which is condensed with allyl oxalyl chloride (XVI) by means of DIEA in CH2Cl2 to give the oxalyl amide (XVII). Compound (XVII) is then treated with AgNO3 and pyridine in acetonitrile to provide the silver mercaptide (XVIII), which is acylated with tetrahydrofuran-2(R)-carbonyl chloride (IV) in acetonitrile to afford the penem precursor (XIX). Finally, compound (XV) is obtained by intramolecular Wittig cyclization of (XX) with P(OEt)3 in refluxing xylene.
Hydrolysis of faropenem allyl ester (XIII) to faropenem sodium (XX) can be performed under several different conditions: i) triphenylphosphine, sodium 2-ethylhexanoate and palladium tetrakis(triphenylphosphine); ii) palladium tetrakis(triphenylphosphine) and sodium 4-(methoxycarbonyl)- 5,5-dimethylcyclohexane-1,3-dione enolate in several different solvents such as methyl acetate, ethyl acetate, tetrahydrofuran, dioxane, sec-butanol, acetonitrile, acetone, 2-butanone, 1,2-dichloroethane, chlorobenzene, toluene, or ethylene glycol dimethyl ether; iii) triphenylphosphine and palladium tetrakis(triphenylphosphine) with sodium propionate, sodium acetate or sodium lactate in tetrahydrofuran or acetone; and iv) palladium acetate in the presence of P(OBu)3 and sodium propionate in THF. Finally, faropenem daloxate can be directly obtained from faropenem sodium (XX) by esterification with 4-(iodomethyl)-5-methyl-1,3-dioxol-2-one (XXI) in DMF.
PATENT
https://patents.google.com/patent/CN103059046A/enFaropenem (Faropenem), chemistry (5R, 6S)-6-[(1R)-hydroxyethyl by name]-2-[(2R)-and tetrahydrofuran (THF)] penem-3-carboxylic acid list sodium salt, by the first exploitation listing in 1997 years of Japanese Suntory company.This medicine is a kind of atypical beta-lactam penems antibiotics, has very strong anti-microbial activity, especially to the anti-microbial activities of the anerobes such as the gram positive organisms such as golden Portugal bacterium, penicillin-fast streptococcus pneumoniae, streptococcus faecium and bacteroides fragilis apparently higher than existing cynnematin, anti-gram-negative bacteria is active similar to oral cephalosporin, and is stable to various β-lactamases.Various clinical studyes show that this medical instrument has clinical effectiveness good, safe, the advantage that renal toxicity and neurotoxicity are little.Its structural formula is as follows: For synthesizing of Faropenem, existing many reports in the prior art, for example CN101125857A has reported following synthetic route: Take (3R, 4R)-3-[(R)-1-tert-butyl dimethyl silica ethyl]-4-[(R)-and acetoxyl group] nitrogen heterocyclic din-2-ketone is as starting raw material, and warp gets intermediate compound I with R-(+)-sulfo-tetrahydrofuran (THF)-2-formic acid condensation; Intermediate compound I is carried out acylation reaction with monoene propoxy-oxalyl chloride under the catalysis of alkali, get intermediate II; Intermediate II cyclization under the effect of triethyl-phosphite gets intermediate III; Intermediate III is sloughed hydroxyl protecting group through the effect of tetrabutylammonium, gets intermediate compound IV; Intermediate compound IV decarboxylize protecting group under [four (triphenylphosphine)] palladium and triphenylphosphine effect gets Faropenem.Find that after deliberation the method for the present synthetic Faropenem of reporting is all similar with the disclosed method of above-mentioned CN101125857A, all need remove in two steps the protecting group of hydroxyl and carboxyl, reaction scheme is longer.When removing above-mentioned protecting group, need to use a large amount of tetrabutylammonium and [four (triphenylphosphine)] palladium and triphenylphosphine; these reagent costs are high, toxicity is large; be unfavorable for large industrial production; and can introduce the heavy metal palladium; so that the heavy metal remnants in the Faropenem exceed standard, be not suitable for the production of bulk drug.And when adopting aforesaid method deprotection base, the yield in per step only can reach 60%-75%, has further increased production cost.Embodiment 6The preparation of FaropenemWith intermediate 3(364.5g, 0.8mol) use the 700mL acetic acid ethyl dissolution, to open and stir, 0 ℃ of lower dropping with the 36g trifluoroacetic acid after the dilution of 100mL ethyl acetate dripped off in 1 hour, 0 ℃ of lower reaction 2h that continues.Stopped reaction stirs the sodium bicarbonate aqueous solution of lower dropping 5%, until reaction solution pH is neutral.Emit water layer from the reactor lower end, discard.In reactor, add gradually the ethanolic soln of sodium bicarbonate, until till no longer including solid and separating out.Suction filtration, filter cake gets white solid powder 230g(productive rate 93.7% with acetone-water (10:3, v/v) recrystallization), M.P. 163-164 ℃, detect through HPLC, purity is 99.8%Reference examples 1(5R, 6S)-6-[(R)-1-hydroxyethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] preparation of penem-3-carboxylic acid propyleneWith (5R, 6S)-6-[(R)-the 1-tert-butyl dimethyl silica ethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] penem-3-carboxylic acid propylene (150g, 0.342mol) and ammonium bifluoride (59.5g, 1.025mmol) add successively among the 400mL DMF, 55~60 ℃ were reacted 5 hours, stopped reaction, suction filtration, filtrate adds water 800ml, uses ethyl acetate extraction, and organic phase is washed with 5% sodium hydrogen carbonate solution, anhydrous sodium sulfate drying, concentrated, gained incarnadine oily matter gets yellow solid 73g through the petrol ether/ethyl acetate recrystallization, yield 66%.Reference examples 2The preparation of Faropenem(the 5R that reference examples 1 is prepared, 6S)-6-[(R)-the 1-hydroxyethyl]-2-[(R)-and the 2-TETRAHYDROFUROYL sulfenyl] penem-3-carboxylic acid propylene (73g, 0.224mol), 6.5g triphenylphosphine, 6.5g [four (triphenylphosphine)] palladium adds among the 500mL methylene dichloride l successively, the ethyl acetate solution that adds the 2 ethyl hexanoic acid sodium preparation of 500mL 0.5M, stirring at room 1 hour, stopped reaction adds 15mL water in reaction solution, stir 30min, suction filtration, this solid is dissolved in the 100mL water again, adds decolorizing with activated carbon 30min, filter, filtrate adds in the 500mL acetone, place crystallization, get Faropenem 66g, yield 96%.Find that by contrast the total recovery that two steps of reference examples remove hydroxyl and carboxyl-protecting group only has about 63.4%, and single stage method of the present invention removes the yield of hydroxyl and carboxyl-protecting group and can reach more than 90%.Preparation method of the present invention can the one-step removal hydroxyl and carboxyl on protecting group, shortened the production cycle, the deprotecting regent cost is low, toxicity is little, can not cause heavy metal remaining, and have higher reaction yield, is fit to very much the industrial production of raw material medicine.
Patent
Publication numberPriority datePublication dateAssigneeTitleCN1939924A *2006-09-082007-04-04鲁南制药集团股份有限公司Industrial production of Fallopeinan sodiumWO2008035153A2 *2006-08-022008-03-27Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of beta-lactam antibioticCN103059046A *2013-01-282013-04-24苏州二叶制药有限公司Preparation method of faropenemFamily To Family CitationsCN100522975C *2007-08-232009-08-05东北制药集团公司沈阳第一制药厂Method for preparing faropenemPublication numberPriority datePublication dateAssigneeTitleCN1884284A *2005-06-212006-12-27浙江金华康恩贝生物制药有限公司Process for the preparation of sodium faropenemCN1939924A *2006-09-082007-04-04鲁南制药集团股份有限公司Industrial production of Fallopeinan sodiumCN101125857A *2007-08-232008-02-20东北制药集团公司沈阳第一制药厂Method for preparing faropenemWO2008035153A2 *2006-08-022008-03-27Orchid Chemicals & Pharmaceuticals LimitedProcess for the preparation of beta-lactam antibiotic
EP0410727A1 *1989-07-261991-01-30Suntory LimitedProcesses for removing allyl groupsUS4997829A *1985-03-091991-03-05Suntory LimitedPenem compounds, and use thereofEP0574940A1 *1992-06-181993-12-22Tanabe Seiyaku Co., Ltd.Method for removing the protecting group for carboxyl groupWO2007039885A1 *2005-10-052007-04-12Ranbaxy Laboratories LimitedA process for the preparation of faropenemFamily To Family Citations Publication numberPriority datePublication dateAssigneeTitleCN102964357A *2012-11-112013-03-13苏州二叶制药有限公司Faropenem sodium and tablet thereofCN103059046A *2013-01-282013-04-24苏州二叶制药有限公司Preparation method of faropenemCN103880864A *2014-03-252014-06-25江苏正大清江制药有限公司Method for synthesizing faropenem sodiumCN104086516A *2014-07-182014-10-08成都樵枫科技发展有限公司Synthetic method of R-(+)-sulfotetrahydrofuran-2-formic acidCN101941981B *2009-07-032015-01-21湖南华纳大药厂有限公司Catalyst composition and method for preparing faropenem sodiumCN106860405A *2015-12-142017-06-20山东新时代药业有限公司A kind of faropenem sodium granules and preparation method thereofCN108840877A *2018-06-122018-11-20赤峰迪生药业有限责任公司A kind of preparation method of oxygen cephalosporin intermediate
2-propylpentanoic acid, DIVALPROEX SODIUM, 76584-70-8, Valproate semisodium, Epival, Depakote, Sodium divalproate, Semisodium Valproate, Abbott 50711, ValdisovalValproic Acid CAS Registry Number: 99-66-1 CAS Name: 2-Propylpentanoic acid Additional Names: 2-propylvaleric acid; di-n-propylacetic acid Trademarks: Convulex (Pharmacia); Depakene (Abbott) Molecular Formula: C8H16O2 Molecular Weight: 144.21 Percent Composition: C 66.63%, H 11.18%, O 22.19% Literature References: Antiepileptic; increases levels of g-aminobutyric acid (GABA) in the brain. Prepn: B. S. Burton, Am. Chem. J.3, 385 (1882); E. Oberreit, Ber.29, 1998 (1896); M. Tiffeneau, Y. Deux, Compte Rend.212, 105 (1941). Anticonvulsant activity: H. Meunier et al.,Therapie18, 435 (1963). Toxicity data: Jenner et al.,Food Cosmet. Toxicol.2, 327 (1964). Comprehensive description: Z. L. Chang, Anal. Profiles Drug Subs.8, 529-556 (1979). Review of teratogenicity studies: H. Nau et al.,Pharmacol. Toxicol.69, 310-321 (1991); R. Alsdorf, D. F. Wyszynski, Expert Opin. Drug Safety4, 345-353 (2005). Review of pharmacology and clinical experience in epilepsy: E. M. Rimmer, A. Richens, Pharmacotherapy5, 171-184 (1985); in psychiatric disease: D. R. P. Guay, ibid.15, 631-647 (1995); in migraine prophylaxis: C. E. Shelton, J. F. Connelly, Ann. Pharmacother.30, 865-866 (1996). Review of pharmacodynamics and mechanisms of action: W. Löscher, Prog. Neurobiol.58, 31-59 (1999). Properties: Colorless liquid with characteristic odor. bp 219.5°. nD24.5 1.425. d40 0.9215. pKa 4.6. Very sol in organic solvents. Soly in water: 1.3 mg/ml. LD50 orally in rats: 670 mg/kg (Jenner). Boiling point: bp 219.5° pKa: pKa 4.6 Index of refraction:nD24.5 1.425 Density: d40 0.9215 Toxicity data: LD50 orally in rats: 670 mg/kg (Jenner) Derivative Type: Sodium salt (1:1) CAS Registry Number: 1069-66-5 Additional Names: Sodium valproate Trademarks: Depacon (Abbott); Depakin (Sanofi-Synthelabo); Dépakine (Sanofi-Aventis); Epilim (Sanofi-Aventis); Ergenyl (Sanofi-Synthelabo); Leptilan (Dolorgiet); Orfiril (Desitin) Molecular Formula: C8H15NaO2 Molecular Weight: 166.19 Percent Composition: C 57.82%, H 9.10%, Na 13.83%, O 19.25% Properties: White, odorless, crystalline, deliquescent powder. pKa 4.8. Hygroscopic. One gram is sol in 0.4 ml water; 1.5 ml ethanol; 5 ml methanol. Practically insol in common organic solvents. LD50 orally in mice: 1700 mg/kg (Meunier). pKa: pKa 4.8 Toxicity data: LD50 orally in mice: 1700 mg/kg (Meunier) Derivative Type: Sodium salt (2:1) CAS Registry Number: 76584-70-8 Additional Names: Sodium hydrogen bis(2-propylpentanoate); divalproex sodium; valproate semisodium Manufacturers’ Codes: Abbott 50711 Trademarks: Depakote (Abbott); Valcote (Abbott) Molecular Formula: C16H31NaO4 Molecular Weight: 310.40 Percent Composition: C 61.91%, H 10.07%, Na 7.41%, O 20.62% Derivative Type: Magnesium salt Trademarks: Depamag (Sigma-Tau) Molecular Formula: C16H30MgO4 Molecular Weight: 310.71 Percent Composition: C 61.85%, H 9.73%, Mg 7.82%, O 20.60% Therap-Cat: Anticonvulsant; antimanic; antimigraine.Keywords: Anticonvulsant; Antimigraine; Antimanic.
Synthesis Reference
Daniel Aubert, Francis Blanc, Henri Desmolin, Michel Morre, Lucette Sindely, “Valproic acid preparations.” U.S. Patent US5017613, issued January, 1965.
https://patents.google.com/patent/WO2007004238A2/enDivalproex sodium is one of the most widely used epileptic agents presently available in the market. Both the constituents, valproic acid and sodium valproate themselves have also been used for the treatment of epileptic seizures and convulsions. But their utility has remained restricted since valproic acid is a liquid and is difficult to formulate for an oral dosage form whereas sodium valproate is a hygroscopic solid with poor stability characteristics. Divalproex sodium is an oligomer having 1:1 molar ratio of valproic acid and sodium valproate containing 4 to 6 units. The relevant prior art includes US 4,988,731 (’73I) relates to a non-hygroscopic stable sodium hydrogen divalproate oligomer. Its probable structure is shown in Fig 1
Fig 1 – Divalproex sodiumWhere M is a about 2.As can be seen from the displayed structure, one mole each of the valproic acid forms coordinate bonds with the sodium of the sodium valproate molecule, and the valproate ion is ionically bonded to the sodium atom. The structure is thus consistent with the unique characteristic of the compound. However the preferred mode of representing Divalproex sodium is by reference to single compound of the formula{(CH3CH2CH2)2CHCO2} {(CH3CH2CH2)2CHCO2}Na, HThe said patent also describes two alternative processes for the preparation of the oligomer. According to one aspect, the oligomer is produced by dissolving sodium valproate and valproic acid in equimolar amount in acetone and crystallizing from chilled acetone at around O0C. Alternatively Divalproex sodium can be isolated from a two component liquid medium, which includes acetone where in half equivalent of NaOH to the valproic acid present, preferable as a solution in an acetone miscible solvent eg. water. The new compound can be recovered from the liquid phase by evaporating the solvent(s) and, if desired, the new compound can be recrystallized, for instance from acetonitrile or others or the material may be spay-dried, lyophilized or purified by chromatography.US ‘731 claims yield of 90% of theory.Drawbacks of the above mentioned reported methods for the preparation of Divalproex sodium described in US 4988731 are difficult to reproduce on a large scale and provides inconsistent yields and the material obtained is not always free flowing in nature. The process involves the crystallization of a 1:1 mixture of valproic acid and sodium valproate from a chilled solution of acetone, followed by washing with chilled acetone. Divalproex sodium is as such fairly soluble in acetone at temperatures above 1O0C and extreme care has to be. taken while performing washing with chilled acetone as any rise in temperature would lead to the loss of yield. This problem actually comes to the fore while scaling up the process during commercialization since during centrifugation of the large volume the temperature of the mixture rises and acetone has to be cooled below O0C, which require large amount of liquid nitrogen or dry ice. Moreover it was also observed that due to the cooled nature of the solvent, the isolated Divalproex sodium absorbs considerable amount of moisture and therefore requires longer time to dry eventually leading to longer time cycle for the otherwise simple single step process. Also the high moisture content in the recovered acetone makes it unsuitable for reuse. Alternatively, to avoid absorption of water, the centrifugation had to be carried out under a blanket of dry nitrogen gas. These additional infrastructural loads add to input costs eventually making the otherwise single step low cost process becoming uncompetitive and economically unviable.Similarly the other process involves the addition of half molar equivalent of sodium hydroxide dissolved in water to valproic acid and the solvent has to be evaporated to obtain crude product, which has to be recrystallized to get Divalprox of the desired specification. The process is operationally tedious and requires the reduction in the level of water in the reaction mass via evaporation of the solvent followed by re- crystallization from acetonitrile making the process lengthy and economically unviable. There is therefore a need for operationally making this single step process more efficient and high yieldingExample I:To lOOg of Valproic acid with stirring at 20-300C, powdered NaOH ( 13g; half molar) is added & the resulting reaction mixture is stirred at 40-500C for 1 hr. Then acetonitrile(600ml) is added to obtain clear solution at 40-500C and the solution is charcoalized at 40-500C followed by filtration at 40-500C through hyflo-bed. The resultant reaction mixture was stirred at 10-200C for 2-3 hr. The solid , thus obtained, was filtered and product was dried at 40-450C for 10-12 hr. (102.25g, 95%)Example II;To lOOg of Valproic acid with stirring at 20-300C, powdered NaOH (13g; half molar) is added & the resulting reaction mixture is stirred at 30-400C for 1 hr. Then acetone (600ml) is added to obtain clear solution at 30-400C and the material is charcoalized at 30-400C followed by filtration through hyflo-bed. The resultant reaction solution was stirred at -5°C to -1O0C for 2-3 hr. The solid , thus obtained, was filtered and product was dried at 40-450C for 10-12 hr. ( 55g, 51.11%) Example III:To a solution of Valproic acid (10Og) in dichloromethane (200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 30- 400C for 1 hr to get clear solution. Then acetonitrile (600ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (10Og; 93%)Example IV:To a solution of Valproic acid (10Og) in diisopropyl ether(200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (104g; 96.65%)Example V:To a solution of Valproic acid (10Og) in methyl tertiary butyl ether(200ml) at 20- 300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (102g;94.79%)Example VI:To a solution of Valproic acid (10Og) in toluene (200ml) at 20-300C, powdered caustic (13g ; half molar) is added & the reaction mixture is stirred at 40-500C for 1 hr to get clear solution. Then acetonitrile (800ml) is added to it inorder to crystallize the product. The solid, thus obtained, is further stirred at 0-50C for 2-3 hr followed by filtration. The product was dried at 40-450C for 10-12 hr. (101g; 93.87%)Example VII: A mixture of sodium valproate (6Og) and valproic acid (52.04g) was taken in acetonitrile (800ml) and heated at reflux to obtain a clear solution, which was filtered through hyflo-bed to remove suspended particles. Then the solution was stirred at 10- 200C for 2-3 hr. The solid, thus obtained, was filtered and washed with acetonitrile (100ml). The product was dried at 40-450C for 10-12 hr. (105g ; 93.75%)Example VIII;To a solution of valproic acid (10Og) in methanol (200ml) at 20-300C5 caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the methanol was recovered at reduced pressure and acetonitrile (600ml) is added to it with stirring. The reaction mixture was further stirred at 0-50C for 2-3 hr. The solid, thus obtained, is filtered, washed with acetonitrile (100ml) and product was dried at 40-45°C for 10-12 hr.(102g; ~ 95%) Example IX:To a solution of valproic acid (10Og) in methanol (200ml) at 20-300C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the methanol was recovered at reduced pressure and acetone (600ml) is added to it with stirring. The reaction mixture was further stirred at -5°C to -1O0C for 2-3 hr. The solid, thus obtained, is filtered, washed with chilled acetone (100ml) and product was dried at 40-450C for 10-12 hr.(54g; ~ 50.11%)Example X:To a solution of valproic acid (10Og) in ethanol (200ml) at 20-300C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the ethanol was recovered at reduced pressure and acetonitrile (600ml) is added to it with stirring.The reaction mixture was further stirred at 0-50C for 2-3 hr. The solid, thus obtained, is filtered, washed with acetonitrile (100ml) and product was dried at 40-450C for 10-12 hr.(101g; ~ 93.87%)Example XI: To a solution of valproic acid (10Og) in ethanol (200ml) at 20-30°C, caustic (13g; half molar) is added & the reaction mixture is stirred at 20-300C for 1 hr. Then the ethanol was recovered at reduced pressure and acetone (600ml) is added to it with stirring. The reaction mixture was further stirred at -5°C to -100C for 2-3 hr. The solid, thus obtained, is filtered, washed with chilled acetone (100ml) and product was dried at 40-450C for 10-12 hr.(55g; ~ 51%)ADVANTAGES:> The process is high yielding. > The process produces Divalproex sodium with improved flowability.> The process produces Divalproex sodium that is non-hygroscopic and more stable.> The process is industrially feasible, precise, reproducible and does not require sophisticated infrastructure.
Divalproex Sodium is a stable coordination compound comprised of sodium valproate and valproic acid with anticonvulsant and antiepileptic activities. Divalproex dissociates to the valproate ion in the gastrointestinal tract. This agent binds to and inhibits gamma-aminobutyric acid (GABA) transaminase and its anticonvulsant activity may be exerted by increasing brain concentration of GABA and by inhibiting enzymes that catabolize GABA or block the reuptake of GABA into glia and nerve endings. Divalproex may also work by suppressing repetitive neuronal firing through inhibition of voltage-sensitive sodium channels.
Valproate semisodium is a mixture of valproic acid and its sodium salt in a 1:1 molar ratio. It is used for the management and treatment of seizure disorders, mania, and prophylactic treatment of migraine headache. It has a role as an antimanic drug, an anticonvulsant and a GABA agent. It contains a valproic acid and a sodium valproate.
Divalproex sodium, valproate sodium, and valproic acid, are all similar medications that are used by the body as valproic acid. Therefore, the term valproic acid will be used to represent all of these medications in this discussion.
Common side effects of valproate include nausea, vomiting, sleepiness, and dry mouth.[2] Serious side effects can include liver failure, and regular monitoring of liver function tests is therefore recommended.[2] Other serious risks include pancreatitis and an increased suicide risk.[2] Valproate is known to cause serious abnormalities in babies if taken during pregnancy,[2][3] and as such it is not typically recommended for women of childbearing age who have migraines.[2]
Valproate was first made in 1881 and came into medical use in 1962.[7] It is on the World Health Organization’s List of Essential Medicines[8] and is available as a generic medication.[2] It is marketed under the brand names Depakote, among others.[2] In 2018, it was the 131st most commonly prescribed medication in the United States, with more than 5 million prescriptions.[9][10]
There is limited evidence that adding valproate to antipsychotics may be effective for overall response and also for specific symptoms, especially in terms of excitement and aggression. Valproate was associated with a number of adverse events among which sedation and dizziness appeared more frequently than in the control groups.[18]
showOutcomeFindings in wordsFindings in numbersQuality of evidence
Valproate is also used to prevent migraine headaches. Because this medication can be potentially harmful to the fetus, valproate should be considered for those able to become pregnant only after the risks have been discussed.[22]
There is evidence that valproic acid may cause premature growth plate ossification in children and adolescents, resulting in decreased height.[26][27][28][29] Valproic acid can also cause mydriasis, a dilation of the pupils.[30] There is evidence that shows valproic acid may increase the chance of polycystic ovary syndrome (PCOS) in women with epilepsy or bipolar disorder. Studies have shown this risk of PCOS is higher in women with epilepsy compared to those with bipolar disorder.[31] Weight gain is also possible.[32]
Pregnancy
Valproate causes birth defects;[33] exposure during pregnancy is associated with about three times as many major abnormalities as usual, mainly spina bifida with the risks being related to the strength of medication used and use of more than one drug.[34][35] More rarely, with several other defects, including a “valproate syndrome”.[36] Characteristics of this valproate syndrome include facial features that tend to evolve with age, including a triangle-shaped forehead, tall forehead with bifrontal narrowing, epicanthic folds, medial deficiency of eyebrows, flat nasal bridge, broad nasal root, anteverted nares, shallow philtrum, long upper lip and thin vermillion borders, thick lower lip and small downturned mouth.[37] While developmental delay is usually associated with altered physical characteristics (dysmorphic features), this is not always the case.[38]
Children of mothers taking valproate during pregnancy are at risk for lower IQs.[39][40][41] Maternal valproate use during pregnancy increased the probability of autism in the offspring compared to mothers not taking valproate from 1.5% to 4.4%.[42] A 2005 study found rates of autism among children exposed to sodium valproate before birth in the cohort studied were 8.9%.[43] The normal incidence for autism in the general population is estimated at less than one percent.[44] A 2009 study found that the 3-year-old children of pregnant women taking valproate had an IQ nine points lower than that of a well-matched control group. However, further research in older children and adults is needed.[45][46][47]
Sodium valproate has been associated with paroxysmal tonic upgaze of childhood, also known as Ouvrier–Billson syndrome, from childhood or fetal exposure. This condition resolved after discontinuing valproate therapy.[48][49]
Women who intend to become pregnant should switch to a different medication if possible or decrease their dose of valproate.[50] Women who become pregnant while taking valproate should be warned that it causes birth defects and cognitive impairment in the newborn, especially at high doses (although valproate is sometimes the only drug that can control seizures, and seizures in pregnancy could have worse outcomes for the fetus than exposure to valproate). Studies have shown that taking folic acid supplements can reduce the risk of congenital neural tube defects.[22] The use of valproate for migraine or bipolar disorder during pregnancy is contraindicated in the European Union, and the medicines are not recommended for epilepsy during pregnancy unless there is no other effective treatment available.[51]
Elderly
Valproate in elderly people with dementia caused increased sleepiness. More people stopped the medication for this reason. Additional side effects of weight loss and decreased food intake were also associated with one-half of people who become sleepy.[22]
Excessive amounts of valproic acid can result in sleepiness, tremor, stupor, respiratory depression, coma, metabolic acidosis, and death.[54] In general, serum or plasma valproic acid concentrations are in a range of 20–100 mg/l during controlled therapy, but may reach 150–1500 mg/l following acute poisoning. Monitoring of the serum level is often accomplished using commercial immunoassay techniques, although some laboratories employ gas or liquid chromatography.[55] In contrast to other antiepileptic drugs, at present there is little favorable evidence for salivary therapeutic drug monitoring. Salivary levels of valproic acid correlate poorly with serum levels, partly due to valproate’s weak acid property (pKa of 4.9).[56]
Valproate inhibits CYP2C9, glucuronyl transferase, and epoxide hydrolase and is highly protein bound and hence may interact with drugs that are substrates for any of these enzymes or are highly protein bound themselves.[24] It may also potentiate the CNS depressant effects of alcohol.[24] It should not be given in conjunction with other antiepileptics due to the potential for reduced clearance of other antiepileptics (including carbamazepine, lamotrigine, phenytoin and phenobarbitone) and itself.[24] It may also interact with:[22][24][62]
Aspirin: may increase valproate concentrations. May also interfere with valproate’s metabolism.
Benzodiazepines: may cause CNS depression and there are possible pharmacokinetic interactions.
Carbapenem antibiotics: reduce valproate levels, potentially leading to seizures.
Cimetidine: inhibits valproate’s metabolism in the liver, leading to increased valproate concentrations.
Erythromycin: inhibits valproate’s metabolism in the liver, leading to increased valproate concentrations.
Ethosuximide: valproate may increase ethosuximide concentrations and lead to toxicity.
Felbamate: may increase plasma concentrations of valproate.
Mefloquine: may increase valproate metabolism combined with the direct epileptogenic effects of mefloquine.
Primidone: may accelerate metabolism of valproate, leading to a decline of serum levels and potential breakthrough seizure.
Rifampicin: increases the clearance of valproate, leading to decreased valproate concentrations
Warfarin: valproate may increase free warfarin concentration and prolong bleeding time.
Zidovudine: valproate may increase zidovudine serum concentration and lead to toxicity.
Pharmacology
Pharmacodynamics
Although the mechanism of action of valproate is not fully understood,[24] traditionally, its anticonvulsant effect has been attributed to the blockade of voltage-gated sodium channels and increased brain levels of gamma-aminobutyric acid (GABA).[24] The GABAergic effect is also believed to contribute towards the anti-manic properties of valproate.[24] In animals, sodium valproate raises cerebral and cerebellar levels of the inhibitory synaptic neurotransmitter, GABA, possibly by inhibiting GABA degradative enzymes, such as GABA transaminase, succinate-semialdehyde dehydrogenase and by inhibiting the re-uptake of GABA by neuronal cells.[24]
Prevention of neurotransmitter-induced hyperexcitability of nerve cells, via Kv7.2 channel and AKAP5, may also contribute to its mechanism.[63] Also, it has been shown to protect against a seizure-induced reduction in phosphatidylinositol (3,4,5)-trisphosphate (PIP3) as a potential therapeutic mechanism.[64]
It also has histone-deacetylase-inhibiting effects. The inhibition of histone deacetylase, by promoting more transcriptionally active chromatin structures, likely presents the epigenetic mechanism for regulation of many of the neuroprotective effects attributed to valproic acid. Intermediate molecules mediating these effects include VEGF, BDNF, and GDNF.[65][66]
Taken by mouth, valproate is rapidly and virtually completely absorbed from the gut.[71] When in the bloodstream, 80–90% of the substance are bound to plasma proteins, mainly albumin. Protein binding is saturable: it decreases with increasing valproate concentration, low albumin concentrations, the patient’s age, additional use of other drugs such as aspirin, as well as liver and kidney impairment.[73][74] Concentrations in the cerebrospinal fluid and in breast milk are 1 to 10% of blood plasma concentrations.[71]
In adult patients taking valproate alone, 30–50% of an administered dose is excreted in urine as the glucuronide conjugate.[75] The other major pathway in the metabolism of valproate is mitochondrial beta oxidation, which typically accounts for over 40% of an administered dose.[75] Typically, less than 20% of an administered dose is eliminated by other oxidative mechanisms.[75] Less than 3% of an administered dose of valproate is excreted unchanged (i.e., as valproate) in urine.[75] Only a small amount is excreted via the faeces.[71]Elimination half-life is 16±3 hours and can decrease to 4–9 hours when combined with enzyme inducers.[71][74]
Valproic acid was first synthesized in 1882 by Beverly S. Burton as an analogue of valeric acid, found naturally in valerian.[76] Valproic acid is a carboxylic acid, a clear liquid at room temperature. For many decades, its only use was in laboratories as a “metabolically inert” solvent for organic compounds. In 1962, the French researcher Pierre Eymard serendipitously discovered the anticonvulsant properties of valproic acid while using it as a vehicle for a number of other compounds that were being screened for antiseizure activity. He found it prevented pentylenetetrazol-induced convulsions in laboratory rats.[77] It was approved as an antiepileptic drug in 1967 in France and has become the most widely prescribed antiepileptic drug worldwide.[78] Valproic acid has also been used for migraine prophylaxis and bipolar disorder.[79]
Limited (depends on the seizure type; it can help with certain kinds of seizures: drug-resistant epilepsy, partial and absence seizures, can be used against glioblastoma and other tumors both to improve survival and treat seizures, and against tonic–clonic seizures and status epilepticus).[81][82][83][84]
Weak evidence. Not recommended for agitation in people with dementia.[90] Increased rate of adverse effects, including a risk of serious adverse effects.[90]
In 2012, pharmaceutical company Abbott paid $1.6 billion in fines to US federal and state governments for illegal promotion of off-label uses for Depakote, including the sedation of elderly nursing home residents.[110][111]
Valproate exists in two main molecular variants: sodium valproate and valproic acid without sodium (often implied by simply valproate). A mixture between these two is termed semisodium valproate. It is unclear whether there is any difference in efficacy between these variants, except from the fact that about 10% more mass of sodium valproate is needed than valproic acid without sodium to compensate for the sodium itself.[114]
Depalept and Depalept Chrono (extended release tablets) are equivalent to Epilim and Epilim Chrono above. Manufactured and distributed by Sanofi-Aventis.
Mexico – Epival and Epival ER (extended release) by Abbott Laboratories
United Kingdom – Depakote (for psychiatric conditions) and Epilim (for epilepsy) by Sanofi-Aventis and generics
United States – Depakote and Depakote ER (extended release) by Abbott Laboratories and generics[22]
India – Valance and Valance OD by Abbott Healthcare Pvt Ltd, Divalid ER by Linux laboratories Pvt Ltd, Valex ER by Sigmund Promedica, Dicorate by Sun Pharma
Germany – Ergenyl Chrono by Sanofi-Aventis and generics
Chile – Valcote and Valcote ER by Abbott Laboratories
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Jump up to:abc Rossi, S, ed. (2013). Australian Medicines Handbook(2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN978-0-9805790-9-3.
^ Olsen KB, Taubøll E, Gjerstad L (2007). “Valproate is an effective, well-tolerated drug for treatment of status epilepticus/serial attacks in adults”. Acta Neurol. Scand. Suppl. 187: 51–4. doi:10.1111/j.1600-0404.2007.00847.x. PMID17419829. S2CID11159645.
^ Guo CY, Ronen GM, Atkinson SA (2002). “Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy”. Epilepsia. 42 (9): 1141–7. doi:10.1046/j.1528-1157.2001.416800.x. PMID11580761. S2CID25499280.
^ Guo CY, Ronen GM, Atkinson SA (2002). “Long-term valproate and lamotrigine treatment may be a marker for reduced growth and bone mass in children with epilepsy”. Epilepsia. 42 (9): 1141–7. doi:10.1046/j.1528-1157.2001.416800.x. PMID11580761. S2CID25499280.
^ Bilo, Leonilda; Meo, Roberta (October 2008). “Polycystic ovary syndrome in women using valproate: a review”. Gynecological Endocrinology. 24 (10): 562–70. doi:10.1080/09513590802288259. PMID19012099. S2CID36426338.
^ Koch S, Göpfert-Geyer I, Jäger-Roman E, et al. (February 1983). “[Anti-epileptic agents during pregnancy. A prospective study on the course of pregnancy, malformations and child development]”. Dtsch. Med. Wochenschr. (in German). 108 (7): 250–7. doi:10.1055/s-2008-1069536. PMID6402356.
^ Umur AS, Selcuki M, Bursali A, Umur N, Kara B, Vatansever HS, Duransoy YK (2012). “Simultaneous folate intake may prevent adverse effect of valproic acid on neurulating nervous system”. Childs Nerv Syst. 28 (5): 729–737. doi:10.1007/s00381-011-1673-9. PMID22246336. S2CID20344828.
^ Luat, AF (20 September 2007). “Paroxysmal tonic upgaze of childhood with co-existent absence epilepsy”. Epileptic Disorders. 9(3): 332–6. doi:10.1684/epd.2007.0119 (inactive 31 May 2021). PMID17884759.
^ Mock CM, Schwetschenau KH (2012). “Levocarnitine for valproic-acid-induced hyperammonemic encephalopathy”. Am J Health Syst Pharm. 69 (1): 35–39. doi:10.2146/ajhp110049. PMID22180549.
^ Matsuoka M, Igisu H (1993). “Comparison of the effects of L-carnitine, D-carnitine and acetyl-L-carnitine on the neurotoxicity of ammonia”. Biochem. Pharmacol. 46 (1): 159–164. doi:10.1016/0006-2952(93)90360-9. PMID8347126.
^ Jump up to:abcdef Haberfeld H, ed. (2021). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Depakine chrono retard 300 mg Filmtabletten.
^ Kumar, S.; Wong, H.; Yeung, S. A.; Riggs, K. W.; Abbott, F. S.; Rurak, D. W. (2000). “Disposition of valproic acid in maternal, fetal, and newborn sheep. II: Metabolism and renal elimination”. Drug Metabolism and Disposition: The Biological Fate of Chemicals. 28(7): 857–864. PMID10859160.
^ Schneemann; Young, Lloyd; Koda-Kimble, Mary Anne, eds. (2001). Angewandte Arzneimitteltherapie (in German). Springer. pp. 28–29. ISBN3540413561.
^ Vasudev K, Mead A, Macritchie K, Young AH (2012). “Valproate in acute mania: is our practice evidence based?”. Int J Health Care Qual Assur. 25 (1): 41–52. doi:10.1108/09526861211192395. PMID22455007.
^ Bond DJ, Lam RW, Yatham LN (2010). “Divalproex sodium versus placebo in the treatment of acute bipolar depression: a systematic review and meta-analysis”. J Affect Disord. 124 (3): 228–334. doi:10.1016/j.jad.2009.11.008. PMID20044142.
^ Candelaria M, Herrera A, Labardini J, González-Fierro A, Trejo-Becerril C, Taja-Chayeb L, Pérez-Cárdenas E, de la Cruz-Hernández E, Arias-Bofill D, Vidal S, Cervera E, Dueñas-Gonzalez A (2011). “Hydralazine and magnesium valproate as epigenetic treatment for myelodysplastic syndrome. Preliminary results of a phase-II trial”. Ann. Hematol. 90 (4): 379–387. doi:10.1007/s00277-010-1090-2. PMID20922525. S2CID13437134.
^ Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, Romanski A, Kramer OH, Kampfmann M, Hoelzer D, Neubauer A, Ruthardt M, Ottmann OG (2005). “Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia”. Cancer. 104 (12): 2717–2725. doi:10.1002/cncr.21589. PMID16294345. S2CID1802132.
^ Kuendgen A, Schmid M, Schlenk R, Knipp S, Hildebrandt B, Steidl C, Germing U, Haas R, Dohner H, Gattermann N (2006). “The histone deacetylase (HDAC) inhibitor valproic acid as monotherapy or in combination with all-trans retinoic acid in patients with acute myeloid leukemia”. Cancer. 106 (1): 112–119. doi:10.1002/cncr.21552. PMID16323176. S2CID43747497.
^ Coronel J, Cetina L, Pacheco I, Trejo-Becerril C, González-Fierro A, de la Cruz-Hernandez E, Perez-Cardenas E, Taja-Chayeb L, Arias-Bofill D, Candelaria M, Vidal S, Dueñas-González A (2011). “A double-blind, placebo-controlled, randomized phase III trial of chemotherapy plus epigenetic therapy with hydralazine valproate for advanced cervical cancer. Preliminary results”. Med. Oncol. 28 Suppl 1: S540–6. doi:10.1007/s12032-010-9700-3. PMID20931299. S2CID207372333.
^ Hicks CW, Pandya MM, Itin I, Fernandez HH (2011). “Valproate for the treatment of medication-induced impulse-control disorders in three patients with Parkinson’s disease”. Parkinsonism Relat. Disord. 17 (5): 379–81. doi:10.1016/j.parkreldis.2011.03.003. PMID21459656.
^ Sriram A, Ward HE, Hassan A, Iyer S, Foote KD, Rodriguez RL, McFarland NR, Okun MS (2013). “Valproate as a treatment for dopamine dysregulation syndrome (DDS) in Parkinson’s disease”. J. Neurol. 260 (2): 521–7. doi:10.1007/s00415-012-6669-1. PMID23007193. S2CID21544457.
Publication numberPriority datePublication dateAssigneeTitleCA1144558A *1979-10-221983-04-12Francis E. FischerProcess for making sodium hydrogen divalproateUS4988731A *1979-08-201991-01-29Abbott LaboratoriesSodium hydrogen divalproate oligomerUS5212326A *1979-08-201993-05-18Abbott LaboratoriesSodium hydrogen divalproate oligomerWO2001032595A1 *1999-11-022001-05-10Cilag AgMethod for producing compounds of the valproinic acidUS20030018215A1 *2001-06-292003-01-23Procos S.P.A.Process for the preparation of sodium divalproatePublication numberPriority datePublication dateAssigneeTitleUS20110040122A1 *2009-08-112011-02-17Sci Pharmtech, Inc.Method for preparing metal salt of valproic acidCN102942467A *2012-10-172013-02-27山东方明药业集团股份有限公司Preparation method of divalproex sodiumCN103183600A *2011-12-302013-07-03北大方正集团有限公司Method for preparing divalproex sodium
CAS Registry Number: 137-58-6 CAS Name: 2-(Diethylamino)-N-(2,6-dimethylphenyl)acetamide Additional Names: 2-diethylamino-2¢,6¢-acetoxylidide; w-diethylamino-2,6-dimethylacetanilide; lignocaine Trademarks: Cuivasil (IDC); Lidoderm (Hind); LidoPosterine (Kade); Vagisil (Combe) Molecular Formula: C14H22N2O Molecular Weight: 234.34 Percent Composition: C 71.75%, H 9.46%, N 11.95%, O 6.83% Literature References: Long-acting, membrane stabilizing agent against ventricular arrhythmia. Originally developed as a local anesthetic. Prepn: N. M. Löfgren, B. J. Lundqvist, US2441498 (1948 to Astra); A. D. H. Self, A. P. T. Easson, GB706409 (1954 to May & Baker); I. P. S. Hardie, E. S. Stern, GB758224 (1956 to J. F. Macfarlane & Co.); Zhuravlev, Nikolaev, Zh. Obshch. Khim.30, 1155 (1960). Toxicity studies: E. R. Smith, B. R. Duce, J. Pharmacol. Exp. Ther.179, 580 (1971); G. H. Kronberg et al.,J. Med. Chem.16, 739 (1973). Review of pharmacokinetics: N. L. Benowitz, W. Meister, Clin. Pharmacokinet.3, 177 (1978). Review of action as local anesthetic: Löfgren, Studies on Local Anesthetics: Xylocaine, A New Synthetic Drug (Hoeggstroms, Stockholm, 1948); Cooper, Pharm. J.171, 68 (1953). Reviews of anti-arrhythmic agents: J. L. Anderson et al.,Drugs15, 271 (1978); L. H. Opie, Lancet1, 861 (1980); E. Carmeliet, Ann. N.Y. Acad. Sci.427, 1 (1984). Comprehensive description: K. Groningsson et al.,Anal. Profiles Drug Subs.14, 207-243 (1985); M. F. Powell, ibid.15, 761-779 (1986). Review of use in treatment of postherpetic neuralgia: P. S. Davies, B. S. Galer, Drugs64, 937-947 (2004).Properties: Needles from benzene or alcohol, mp 68-69°. bp4 180-182°; bp2 159-160°. Insol in water. Sol in alcohol, ether, benzene, chloroform, oils. Partition coefficient (octanol/water, pH 7.4): 43. Melting point: mp 68-69° Boiling point: bp4 180-182°; bp2 159-160° Log P: Partition coefficient (octanol/water, pH 7.4): 43 Derivative Type: Hydrochloride CAS Registry Number: 73-78-9; 6108-05-0 (monohydrate) Trademarks: Basicaina (Galenica); Batixim (So.Se.); Dynexan (Kreussler); Heweneural (Hevert); Licain (DeltaSelect); Lidesthesin (Ritsert); Lidofast (Angelini); Lidoject (Hexal); Lidrian (Baxter); Odontalg (Giovanardi); Sedagul (Wild); Xylocaine (AstraZeneca); Xylocard (AstraZeneca); Xylocitin (Jenapharm); Xyloneural (Strathmann) Molecular Formula: C14H22N2O.HCl Molecular Weight: 270.80 Percent Composition: C 62.09%, H 8.56%, N 10.34%, O 5.91%, Cl 13.09% Properties: Crystals, mp 127-129°; monohydrate, mp 77-78°. Very sol in water, alcohol; sol in chloroform. Insol in ether. pH of 0.5% aq soln: 4.0-5.5. LD50 in mice (mg/kg): 292 orally (Smith, Duce); 105 i.p.; 19.5 i.v. (Kronberg). Melting point: mp 127-129°; mp 77-78° Toxicity data: LD50 in mice (mg/kg): 292 orally (Smith, Duce); 105 i.p.; 19.5 i.v. (Kronberg) Therap-Cat: Anesthetic (local); antiarrhythmic (class IB). Therap-Cat-Vet: Anesthetic (local). Keywords: Anesthetic (Local); Antiarrhythmic.
https://patents.google.com/patent/CN102070483B/en#:~:text=The%20method%20comprises%20the%20following,as%20solvent%20and%20carbonate%20isPreparation method of the present invention, it can be two-step approach, comprises the steps:1) 2,6-xylidine is dissolved in the acetone, adds carbonate then, the back that stirs drips chloroacetyl chloride, and 20~35 ℃ (room temperature) be stirring reaction 3h down; After-filtration is finished in reaction, and after filter cake was washed with water to filtrate and is neutrality, drying made intermediate chloracetyl-2, the 6-xylidine, and yield is about about 94%; 2) intermediate that step 1) is made is dissolved in the acetone, adds carbonate then, and the back that stirs drips diethylamine, back flow reaction 8h; After-filtration is finished in reaction, and filtrate is recrystallization, drying after removing solvent under reduced pressure, makes lignocaine.Wherein, in the step 1) 2, the mol ratio of 6-xylidine, chloroacetyl chloride and carbonate is 1: 1.2~1.7: 1.3~2.0, is preferably 1: 1.5: 1.6. Step 2) intermediate chloracetyl-2 in, the mol ratio of 6-xylidine, diethylamine and carbonate is 1: 1.5~2.5: 1.2~2.0, is preferably 1: 2: 1.5.In addition, preparation method of the present invention owing to all be that solvent, carbonate are catalyzer with acetone in the two-step reaction, therefore can further optimize reaction process on the basis of two-step approach, namely the intermediate of Sheng Chenging needn’t pass through aftertreatment, prepares lignocaine by one kettle way.Described one kettle way comprises the steps: 2,6-xylidine is dissolved in the acetone, adds carbonate then, after stirring, drips chloroacetyl chloride, and 20~35 ℃ (room temperature) be reaction 3h down; After reaction is finished, without processing, directly drip diethylamine, back flow reaction 8h, after-filtration is finished in reaction, and filtrate is recrystallization, drying after removing solvent under reduced pressure, makes lignocaine.Wherein, described 2, the mol ratio of 6-xylidine, chloroacetyl chloride, diethylamine and carbonate is 1: 1.2~1.7: 1.5~2.5: 2.5~3.5, is preferably 1: 1.5: 2: 2.5. In addition, preparation method of the present invention adopts TLC monitoring reaction progress, and the developping agent of TLC is sherwood oil: ethyl acetate (V/V)=3: 1.The invention has the advantages that, the method synthesis technique for preparing lignocaine of the present invention is simple, do not need in the intermediate aftertreatment first pickling numerous and diverse step of alkali cleaning again, avoided unnecessary loss, therefore the yield of the intermediate that makes of the inventive method and lignocaine is all higher, and the lignocaine purity that makes is good, reaches more than 99%, has favorable industrial application prospect; In addition, the inventive method uses acetone to make solvent, and this solvent is nontoxic substantially non-stimulated, and can recycle, and is environmentally friendly. EmbodimentBelow further specify the present invention by specific embodiment, but be not used for limiting the scope of the invention. Embodiment 1 two-step approach prepares lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineAdd 102g 2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, adds 200g salt of wormwood again, and mechanical stirring evenly back drips 100mL chloroacetyl chloride (1.5h drips off), (20 ℃) stirring reaction 3h under the room temperature; Reaction finishes the back suction filtration, and filter cake is washed with water to filtrate and is neutral, and under 100 ℃ of temperature dry 1 hour then, make the 156g white powder, be intermediate chloracetyl-2,6-xylidine, yield are 94%, fusing point is 145.0~147.0 ℃.2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, and the dissolving back adds 112g salt of wormwood, drips the 60g diethylamine fast, back flow reaction 8h; Reaction finishes the back suction filtration, and filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, uses 150mL sherwood oil recrystallization then, suction filtration, vacuum-drying 6h under 40 ℃ of temperature makes the 90g white powder, is lignocaine, yield is 95%, and fusing point is 67.0~68.0 ℃, and content is 99.05%. Embodiment 2 two-step approachs prepare lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineAdd 102g 2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, adds 163g salt of wormwood again, and mechanical stirring evenly back drips 80mL chloroacetyl chloride (1.5h drips off), (20 ℃) stirring reaction 3h under the room temperature; Reaction finishes the back suction filtration, and filter cake is washed with water to filtrate and is neutral, and under 100 ℃ of temperature dry 1 hour then, make the 136g white powder, be intermediate chloracetyl-2,6-xylidine, yield are 82%, fusing point is 145~146 ℃. 2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, and the dissolving back adds 90g salt of wormwood, drips the 45g diethylamine fast, back flow reaction 8h; Reaction finishes the back suction filtration, and filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, uses 150mL sherwood oil recrystallization then, suction filtration, vacuum-drying 6h under 40 ℃ of temperature makes the 84g white powder, is lignocaine, yield is 89%, and fusing point is 66~67 ℃, and content is 99.15%. Embodiment 3 two-step approachs prepare lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineAdd 102g 2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, adds 250g salt of wormwood again, and mechanical stirring evenly back drips 113mL chloroacetyl chloride (1.5h drips off), (20 ℃) stirring reaction 3h under the room temperature; Reaction finishes the back suction filtration, and filter cake is washed with water to filtrate and is neutral, and under 100 ℃ of temperature dry 1 hour then, make the 150g white powder, be intermediate chloracetyl-2,6-xylidine, yield are 90%, fusing point is 147~148 ℃. 2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL acetone, and the dissolving back adds 150g salt of wormwood, drips the 75g diethylamine fast, back flow reaction 8h; Reaction finishes the back suction filtration, and filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, uses 150mL sherwood oil recrystallization then, suction filtration, vacuum-drying 6h under 40 ℃ of temperature makes the 88g white powder, is lignocaine, yield is 93%, and fusing point is 68~69 ℃, and content is 98.75%. Embodiment 4 one kettle ways prepare lignocaineIn the 5000mL there-necked flask, add 305g 2, the 6-xylidine, make solvent with 2000mL acetone, add 700g salt of wormwood again, mechanical stirring evenly back slowly drips 230mL chloroacetyl chloride (1.5h drips off), room temperature (35 ℃) is stirring reaction 3h down, and TLC point plate (use sherwood oil: ethyl acetate (V/V)=3: 1 is made developping agent) demonstration reacts completely; Dropwise 5 50g diethylamine then, the back back flow reaction 8h that stirs, TLC monitoring (developping agent is the same) shows and reacts completely; The reaction solution suction filtration, filtrate is removal of solvent under reduced pressure under 40 ℃ of temperature, gets light yellow solid, uses sherwood oil recrystallization secondary then, makes 482g white lignocaine crystal, and total recovery is 82%, and fusing point is 68.0~69.0 ℃, and content is 99.75%. Comparative example 1 existing method prepares lignocaine1) intermediate chloracetyl-2, the preparation of 6-xylidineIn the 1000mL there-necked flask, with 102g 2, the 6-xylidine is dissolved in the 400mL glacial acetic acid, stirs slowly to add the 100mL chloroacetyl chloride down, is heated to 45 ℃, adds 200g solid sodium acetate (containing crystal water) then, reaction 2h; After reaction finished, ice bath was cooled to below 10 ℃, suction filtration, filter cake is washed with water to filtrate and is neutral, and drying is 1 hour under 100 ℃ of temperature, makes the 111g white powder, be intermediate chloracetyl-2,6-xylidine, yield are that 67% fusing point is 145.0~148.0 ℃. 2) preparation of lignocaineAdd 80g intermediate chloracetyl-2 in the 1000mL there-necked flask, the 6-xylidine is made solvent with 400mL toluene, and the dissolving back drips 60g diethylamine, back flow reaction 3.5h fast; After reaction finished, ice bath was cooled to 5 ℃, suction filtration, filtrate is used the 3mol/L hcl as extraction agent, and the acid solution that extraction is obtained is cooled to 10 ℃ then, stirs slowly to add 6mol/L KOH solution down, be alkalescence (pH8~9) to solution, with pentane extraction, organic layer was after washing, Anhydrous potassium carbonate drying after ice bath was cooled to 20 ℃, vapor bath is steamed and is desolventized, make the 74g white powder, be lignocaine, yield is 78%, fusing point is 66.0~67.0 ℃, and content is 97.15%.Embodiment 1 compares with comparative example 1, its intermediate chloracetyl-2, and the yield of 6-xylidine obviously improves, and reaches 94%, and the total recovery of final product lignocaine also is significantly improved, and the content of lignocaine is brought up to about 99%.Embodiment 4 compares with comparative example 1, and the total recovery of lignocaine is significantly improved, and reach 82%, and content is brought up to more than 99%. Though above with a general description of the specific embodiments, the present invention is described in detail, on basis of the present invention, can make some modifications or improvements it, this will be apparent to those skilled in the art.Therefore, these modifications or improvements all belong to the scope of protection of present invention without departing from theon the basis of the spirit of the present invention. CLIPhttps://www.cerritos.edu/chemistry/chem_212/Documents/Lab/10_lidocaine.pdf
Procedure: (1st week)A: Synthesis of 2,6-Dimethylaniline via Reduction of 2,6-Dimethylnitrobenzene 1. Dissolve1.0 g of 2,6-dimethylnitrobenzene in 10 mL of glacial acetic acid in a 50 mL Erlenmeyer flask. 2. In a 25 mL flask, dissolve 4.6 grams of SnCl2 · 2H2O in 8 mL of concentrated HCl, inside the fume hood. 3. Add the SnCl2 solution in one portion to the nitroxylene solution, magnetically swirl and mix, and let the mixture stand for 15 minutes. 4. Cool the mixture and collect the crystalline salt (dimethylaniline in the salt form: C6H5NH3 +Cl- ) in a Buchner funnel. 5. Transfer the moist crystals to an Erlenmeyer flask, add 5-10 mL of water, and make the solution strongly basic (to remove the acid and change C6H5NH3 +Clback intoC6H5NH2) by adding 30% KOH solution (12 to 17 mL required). 6. After cooling extract with three 10 mL portions of ether, rinse the ether extracts twice with 10 mL of water, and dry over K2CO3. 7. Evaporate the dried and filtered solution to an oil, transfer and rinse into a 50 mL Erlenmeyer flask, complete evaporation, weigh, and calculate the %yield of 2,6-dimethylaniline. B: Synthesis of α-Chloro-2,6-dimethylacetanilide (prepare for a steam bath ahead of time) 1. For every 7 grams (from this step on, you need to calculate proportionally how much you need to add according to the actual weight that you got) of dimethylaniline from the previous step, add 50 mL of glacial acetic acid, and 7.2 g (or 5.2 mL) of chloroacetyl chloride, in that order. 2. Warm the solution on a steam bath to (40–50)ºC, remove, and add a solution of 1 gram of sodium acetate in 100 mL of water. 3. Cool the mixture and collect the product in a Buchner funnel. 4. Transfer the product to a disk of medium–sized filter paper, finely divide it with a spatula, and let air dry until the next laboratory period. 5. Upon drying, measure the mass and the melting point. Also, calculate the % yield. B: Synthesis of α-Chloro-2,6-dimethylacetanilide (prepare for a steam bath ahead of time) 1. For every 7 grams (from this step on, you need to calculate proportionally how much you need to add according to the actual weight that you got) of dimethylaniline from the previous step, add 50 mL of glacial acetic acid, and 7.2 g (or 5.2 mL) of chloroacetyl chloride, in that order. 2. Warm the solution on a steam bath to (40–50)ºC, remove, and add a solution of 1 gram of sodium acetate in 100 mL of water. 3. Cool the mixture and collect the product in a Buchner funnel. 4. Transfer the product to a disk of medium–sized filter paper, finely divide it with a spatula, and let air dry until the next laboratory period. 5. Upon drying, measure the mass and the melting point. Also, calculate the % yield. D. Synthesis of the bisulfate salt of lidocaine 1. Dissolve the lidocaine in ether (10 mL per gram of lidocaine) and add 2 mL of 2.2 M sulfuric acid in ethanol per gram of lidocaine. 2. Stir and scratch with a glass rod to mix and induce crystallization. 3. Dilute the mixture with an equal volume of acetone to aid filtration and collect the salt in a small Buchner funnel. 4. Rinse the solid on the funnel with a few milliliters of acetone and air dry and weigh the product. 5. Calculate the % yield of this step. *** Overall % Yield The overall % YCLIPhttp://home.sandiego.edu/~khuong/chem302L/Handouts/Lidocaine_handout_Su07.pdfSynthetic Strategy Lidocaine will be prepared via a three-step linear synthesis starting from 2,6-dimethylnitrobenzene. The reduction of 2,6-dimethylnitrobenzene 1 with three equivalents of stannous chloride (SnCl2) yields the ammonium salt 2. It is very important that the reaction mixture is strongly acidic during this reaction because the reduction of nitrobenzene using different reducing reagents and conditions can afford a variety of functional groups: nitroso, hydroxylamine (zinc dust, pH 4), azoxy (sodium arsenite), azo (zinc, weakly basic), or hydrazo (zinc, strongly basic). In industrial settings, often iron or tin with hydrochloric acid is used instead of stannous chloride because iron and tin are cheaper, but the reduction takes much longer. In the workup portion of the reaction, the ammonium salt 2 is reacted with an aqueous potassium hydroxide solution, liberating the free 2,6-dimethylaniline 3 in an acid-base reaction.
The reaction of 3 with the bifunctional α-chloroacetyl chloride leads to α-chloro-2,6-dimethylacetanilide 4. A slight excess of the acid chloride is used to ensure the complete conversion of the amine to the amide. The formation of the amide is a result of the significantly higher reactivity (~106 times) of the acyl chloride over the alkyl chloride. The addition of sodium acetate solution avoids the formation of HCl which would protonate unreacted 3 causing it to co-precipitate with the desired product 4.
In the last step, diethylamine performs a nucleophilic substitution (SN2) on the remaining alkyl chloride. Diethylamine serves both as a nucleophile to form lidocaine 5, and as acid scavenger, leading to formation of NH2Et2 + Cl- in this reaction. Since diethylamine is not a very strong nucleophile, it is used in excess here to improve the yield and speed up the reaction. The unreacted amine is later removed by extraction with water. The aqueous extraction of lidocaine with acid separates the unreacted chloroanilide 4 and the lidocaine. After addition of a strong base like aqueous potassium hydroxide, crude lidocaine is obtained.
Procedure Synthesis of 2,6-dimethylaniline (3) Dissolve 15 g of SnCl2•2H2O in 27 mL of concentrated hydrochloric acid. If necessary, heat the mixture gently. Add this solution in one portion to a solution of 3 mL of 2,6-dimethylnitrobenzene in 34 mL of glacial acetic acid. Swirl the resulting mixture and then allow it to stand for 15 minutes before placing the mixture in an ice bath. Collect the formed precipitate by vacuum filtration. Place the wet precipitate obtained above in a beaker and add 20 mL of water. Neutralize the acidic mixture by carefully adding an 8 M aqueous potassium hydroxide with continuous stirring until basic to litmus. Place the mixture in an ice bath. Upon cooling to room temperature, extract the mixture three times with diethyl ether. Combine the organic layers and wash them twice with water and once with brine. Dry the organic layer over anhydrous potassium carbonate. Decant away from the drying agent and evaporate the diethyl ether from a dry, preweighed flask using a rotary evaporator. The oily residue will be your crude product 3. Obtain and record the following information: 1. crude product description (co2. crude weight/percent yieldSynthesis of α-chloro-2,6-dimethylacetanilide (4) Dissolve 3 in 17 mL of glacial acetic acid. Add 1.1 equivalents (based on the moles of 3) of α-chloroacetyl chloride to this solution. Heat the solution to 40-50 o C for ten minutes to complete the reaction. Upon cooling, add a solution of ~3.3 g sodium acetate trihydrate in 67 mL water and then place the resulting mixture in an ice bath. Collect the precipitate by vacuum filtration. Rinse the filter cake with copious amounts of water in order to remove the acetic acid. It is important that the product be completely free of acetic acid after this step (why?). The pH of the individual water rinses can be checked with litmus paper to determine if the product is acid free. Allow for the product to air-dry on a watch glass until the next meeting. There is a reasonable chance that you will not obtain a precipitate as described above. If this is the case, you can try “seeding” using a small sample of authentic product from a classmate. If this does not work, check the TLC to be sure that you have formed product and devise an extractive workup that will separate the unreacted aniline 3 from the desired product 4. (Make sure you understand how to do this even if you obtain a precipitate in the first place). After the aqueous workup and following removal of solvent, you should obtain a solid. If not, check the TLC, using a sample of authentic product from a classmate as a standard. If the product appears relatively pure, you can continue even though the material is not a solid. Obtain and record the following information: 1. crude product description (color, physical state, etc.) 2. crude weight/percent yield 3. mp (if a solid) 4. TLC analysis 5. IR (check for presence of amide functional group) Synthesis of lidocaine; α-(N,N-diethylamino)-2,6-dimethylacetanilide (5). In a round bottom flask, dissolve α-chloro-2,6-dimethylacetanilide 4 in 17 mL of toluene. Before continuing, spot several (4 to 5) TLC plates in advance with this solution of 4. Provide three lanes and spot the 4 on the “SM” and “CO-SPOT” lanes. You will use these plates to monitor the progress of this reaction. Add three equivalents of diethylamine to the round bottom flask, and reflux the mixture vigorously until the reaction is complete. The amount of time required for complete reaction depends on many factors but it will likely take anywhere from more than a few minutes up to several hours. If the reaction is not complete when your lab period ends, you can stopper the reaction and reflux it for additional time at the next period. Usually a white precipitate forms during the reflux. Upon cooling, transfer the reaction mixture to a separatory funnel and extract the mixture three times with water. Next, extract the organic layer with two portions of 3 M hydrochloric acid. Cool the combined acidic aqueous extracts in an ice bath and then add 8 M aqueous potassium hydroxide slowly until the mixture is strongly basic again. The formation of a thin, dark yellow oily layer on top or a white solid is observed at this point. Place the mixture in an ice bath. Once the mixture is chilled, try to initiate the crystallization of the final product if no solid has formed at this point. Collect the obtained precipitate by filtration using a Büchner funnel. Wash it with twice with water and then press it as dry as possible. Obtain and record the following information: 1. crude product description (color, physical state, etc.) 2. crude weight/percent yield 3. TLC analysis Recrystallize the crude product from hexanes. Regardless of the final physical state of your product (solid or oil), obtain and record the following: 1. pure product description (color, physical state, etc.) 2. pure product weight/percent yield 3. overall (three-step) percent yield (from starting material 1) 4. TLC analysis 5. melting point (if a solid) 6. IR 7. 1 H and 13C NMR spectra of lidocaine will be given to you. Turn in a sample of your final product.
1H NMR
13C NMR
MS
IR KBR
Lidocaine is an antiarrhythmic medicine and also serves as a local anaesthetic drug. It is utilized in topical application to relieve pain, burning and itching sensation caused from skin inflammations. This drug is mainly used for minor surgeries. Figure 1 shows the 1H NMR spectrum of 200 mM lidocaine in CDCl3.
Figure 1. Proton NMR spectrum of 200 mM lidocaine in CDCl3.
1H NMR Relaxation
Figures 2, 3 and 4 show the relaxation time measurements. It can be seen that the relaxation times are shortest for the CH2 protons and longest for the CH protons. The first data point amplitude increases with the number of protons for the related peak.
Figure 2. Proton T1 relaxation time measurement of 200 mM lidocaine in CDCl3.
Figure 3. Proton T2 relaxation time measurement of 200 mM lidocaine in CDCl3.
Figure 4. COSY spectrum of 200 mM lidocaine in CDCl3. The cross-peaks and corresponding exchanging protons are labeled by colour-coded arrows and ellipses.
2D COSY
Figure 4 shows the 2D COSY spectrum where two spin systems (6,7,8) to (10,11) can be clearly seen. For instance, the methyl groups at 10 and 11 positions bond to aromatic protons at 6 and 8 positions, while the methyl groups at 16 and 17 positions bond to the ethylene groups at 14 and 15 positions. No coupling occurs at positions (6,7,8) to (16,17) or (14,15).
2D Homonuclear J-Resolved Spectroscopy
The chemical shift in the 2D homonuclear j-resolved spectrum appears along the direct (f2) direction and the effects of coupling between protons appear along the indirect (f1) dimension. This enables the assignment of chemical shifts of multiplets and may help in measuring unresolved couplings. Also, a decoupled 1D proton spectrum is produced by the projection along the f1 dimension. The 2D homonuclear j-resolved spectrum of lidocaine, plus the 1D proton spectrum (blue line) are shown in Figure 5.
Figure 5. Homonuclear j-resolved spectrum of 200 mM lidocaine in CDCl3. The multiplet splitting frequencies for different couplings are colour- coded.
The projection which is vertical reveals how the multiplets disintegrate into a single peak, which makes the 1D spectrum more simplified. Peak multiplicities are produced by vertical traces from peaks in the 2D spectrum and help in determining the frequencies of proton-proton coupling. When coupling frequencies are compared between different peaks, information can be obtained regarding which peaks are bonded to each other. Also, Information regarding the coupling strength can be obtained from the size of the coupling frequencies. These couplings substantiate the results of the COSY experiment.
However, in this experiment, the effects of second order coupling appear in the f1 direction as additional peaks which are equidistant from the coupling partners detached from the zero frequency in the f1 dimension. These peaks provide proof of second order coupling partners, but are generally considered as artifacts. Figure 6 shows these coupling partners and additional peaks marked by colour-coded arrows and ellipses.
Figure 6. Homonuclear j-resolved spectrum of 200 mM lidocaine in CDCl3 showing the extra peaks due to strong couplings.
1D 13C Spectra
Figure 7 shows the 13C NMR spectra of 1 M lidocaine in CDCl3. Since the 1D Carbon experiment is highly susceptible to the 13C nuclei in the specimen, it easily and clearly resolves 9 resonances. In this experiment, only carbons coupled to protons are seen.
Figure 7. Carbon spectra of 1 M lidocaine in CDCl3.
Given the fact that the DEPT spectra do not display the peaks at 170 and 135ppm, they must be part of quaternary carbons. The DEPT-135 and the DEPT-45 experiments provide signals of CH3, CH2 and CH groups, while the DEPT-90 experiment provides only the signal of CH groups. However, in DEPT-135 the CH2 groups occur as negative peaks. It can thus be summed up that the peaks between 45 and 60ppm belong to ethylene groups; the peaks between 10 and 20ppm are part of the methyl groups; and the peaks between 125 and 130ppm belong to methyne groups. A similar study can be carried out on the C and CH peaks.
Heteronuclear Correlation
The Heteronuclear Correlation (HETCOR) experiment identifies the proton signal that appears along the indirect dimension and the carbon signal along the direct dimension. Figure 8 shows the HETCOR spectrum of 1 M lidocaine in CDCl3. in the 2D spectrum, the peaks reveal which proton is attached to which carbon. This experiment helps in resolving assignment uncertainty from the ID carbon spectra.
Figure 8. HETCOR spectrum of 1 M lidocaine in CDCl3.
Heteronuclear Multiple Quantum Coherence
Heteronuclear Multiple Quantum Coherence (HMQC) is similar to the HETCOR experiment and is utilized to associate proton resonances to the carbons that are coupled directly to those protons. But in the HMQC experiment, the proton signal appears along the direct dimension and the carbon signal along the indirect dimension. Figure 9 shows the HMQC spectrum of 1 M lidocaine in CDCl3. In the 2D spectrum, the peaks show which proton is attached to which carbon. For conclusive peak assignment, a similar study with the HETCOR spectrum can be carried out.
Figure 9. HMQC spectrum of 1 M lidocaine in CDCl3.
Heteronuclear Multiple Bond Correlation
The Heteronuclear Multiple Bond Correlation (HMBC) experiment can be employed to achieve long-range correlations of proton and carbon via two or three bond couplings. Similar to the HMQC experiment, the proton signal appears along the direct dimension and the carbon signal along the indirect dimension. Figure 10 shows the HMBC spectrum of 1 M lidocaine in CDCl3.
Figure 10. HMBC spectrum of 1 M lidocaine in CDCl3, with some of the long-range couplings marked.
The couplings amid the molecular positions appear analogous to the couplings seen in the COSY spectrum; however, the HMBC also displays couplings to quaternary carbons, which are not seen either in HMQC or COSY experiments. In addition, there is a correlation between protons and carbons. This is attributed to three-bond bonding from 14 and 15 and vice versa, as shown in light green in Figure 1.
SYN
Synthesis of lidocaine T. J. Reilly (1999). “The Preparation of Lidocaine”. J. Chem. Ed.76 (11): 1557.
CLIP
The Present Synthesis Of Lidocaine Begins With 2,6-Dimethylnitrobenzene (1). This Compound Can Be Made From 1,3-Dimethylbenzene, Also Known As M-Xylene, Which Is More Difficult To Make. Luckily,
This problem has been solved!
See the answer
The present synthesis of lidocaine begins with 2,6-dimethylnitrobenzene (1). This compound can be made from 1,3-dimethylbenzene, also known as m-xylene, which is more difficult to make. Luckily, m-xylene is commercially available, so a synthesis of 1 from m-xylene is a practical alternative if one wants to begin the synthesis of lidocaine with m-xylene. Suppose you want to prepare 1 from m-xylene. Show with chemical equations the reagents that you would use, and the possible isomers that would result.
2. The practical transformation of 1 into 3 is carried out by the following scheme:
Suppose you dissolve the solid precipitate of 2 in water, but forget to include the KOH in the second step above. What would happen after the extraction with ether? Give your answer in terms of what would be found in the ether layer, and in the aqueous layer.
3. Suppose you’re out of acetic acid (CH3COOH) and decide to use ethanol (CH3 CH2OH) as the solvent in the transformation of 3 into 4. Would this be a wise choice, and why?
4.The amide 4 has a nitrogen attached to the benzene ring, and a chlorine attached to a primary carbon. Yet, it doesn’t react with itself in a nucleophilic displacement. Why is the nitrogen in the amide not nucleophilic? Give your answer in terms of the resonance forms of amides in general:
5. In the reaction below, what factors come into play to favor attack of the aniline 3 on the carbonyl carbon of the acid chloride (carbon 1 in red), rather than at the a-carbon (carbon 2 in red)?
6. Before carrying out the transformation below, compound 4 and the glassware used must be oven-dried. What would happen if the reaction was attempted using wet 4?
7.In the reaction below, what factors come into play to favor attack of diethylamine on the a-carbon (carbon 1 in red), rather than on the amide C=O carbon (carbon 2 in red)?
8. In the reaction below, why does the amine nitrogen (#1 in red) undergo protonation with H2SO4 preferentially over the amide nitrogen (#2 in red)? In other words, why is nitrogen 1 basic, but nitrogen 2 is not?
9.Lidocaine and other drugs containing amino groups are usually marketed as their hydrochloride or hydrogen sulfate salts, rather than as “free amines.” Provide two reasons why this practice makes sense.
10.Although lidocaine is marketed as its hydrochloride salt, it doesn’t exhibit the same level of physiological activity as the free amine. The free amine is more lipophilic and diffuses across a neuron cell membrane more rapidly than the ionic salt, resulting in a more rapid onset of anesthesia. Therefore, sodium bicarbonate (NaHCO3) is added to a solution of lidocaine prior to injection. How does the addition of sodium bicarbonate promote a faster anesthetic effect?
CLIP
CLIP
Lidocaine, also known as lignocaine and sold under the brand name Xylocaine among others, is a local anesthetic of the aminoamide type. It is also used to treat ventricular tachycardia.[7][8] When used for local anaesthesia or in nerve blocks, lidocaine typically begins working within several minutes and lasts for half an hour to three hours.[8][9] Lidocaine mixtures may also be applied directly to the skin or mucous membranes to numb the area.[8] It is often used mixed with a small amount of adrenaline (epinephrine) to prolong its local effects and to decrease bleeding.[8]
If injected intravenously, it may cause cerebral effects such as confusion, changes in vision, numbness, tingling, and vomiting.[7] It can cause low blood pressure and an irregular heart rate.[7] There are concerns that injecting it into a joint can cause problems with the cartilage.[8] It appears to be generally safe for use in pregnancy.[7] A lower dose may be required in those with liver problems.[7] It is generally safe to use in those allergic to tetracaine or benzocaine.[8] Lidocaine is an antiarrhythmic medication of the class Ib type.[7] This means it works by blocking sodium channels and thus decreasing the rate of contractions of the heart.[7] When injected near nerves, the nerves cannot conduct signals to or from the brain.[8]
Lidocaine was discovered in 1946 and went on sale in 1948.[10] It is on the World Health Organization’s List of Essential Medicines.[11] It is available as a generic medication.[8][12] In 2018, it was the 233rd most commonly prescribed medication in the United States, with more than 2 million prescriptions.[13][14]
Medical uses
Local numbing agent
The efficacy profile of lidocaine as a local anaesthetic is characterized by a rapid onset of action and intermediate duration of efficacy. Therefore, lidocaine is suitable for infiltration, block, and surface anaesthesia. Longer-acting substances such as bupivacaine are sometimes given preference for spinal and epidural anaesthesias; lidocaine, though, has the advantage of a rapid onset of action. Adrenaline vasoconstricts arteries, reducing bleeding and also delaying the resorption of lidocaine, almost doubling the duration of anaesthesia.
Lidocaine is one of the most commonly used local anaesthetics in dentistry. It can be administered in multiple ways, most often as a nerve block or infiltration, depending on the type of treatment carried out and the area of the mouth worked on.[15]
For surface anaesthesia, several formulations can be used for endoscopies, before intubations, etc. Buffering the pH of lidocaine makes local numbing less painful.[16] Lidocaine drops can be used on the eyes for short ophthalmic procedures. There is tentative evidence for topical lidocaine for neuropathic pain and skin graft donor site pain.[17][18] As a local numbing agent, it is used for the treatment of premature ejaculation.[19]
An adhesive transdermal patch containing a 5% concentration of lidocaine in a hydrogel bandage, is approved by the US FDA for reducing nerve pain caused by shingles.[20] The transdermal patch is also used for pain from other causes, such as compressed nerves and persistent nerve pain after some surgeries.
A 2013 review on treatment for neonatal seizures recommended intravenous lidocaine as a second-line treatment, if phenobarbital fails to stop seizures.[22]
Other
Intravenous lidocaine infusions are also used to treat chronic pain and acute surgical pain as an opiate sparing technique. The quality of evidence for this use is poor so it is difficult to compare it to placebo or an epidural.[23]
Inhaled lidocaine can be used as a cough suppressor acting peripherally to reduce the cough reflex. This application can be implemented as a safety and comfort measure for patients who have to be intubated, as it reduces the incidence of coughing and any tracheal damage it might cause when emerging from anaesthesia.[24]
For gastritis, drinking a viscous lidocaine formulation may help with the pain.[27]
Adverse effects
Adverse drug reactions (ADRs) are rare when lidocaine is used as a local anesthetic and is administered correctly. Most ADRs associated with lidocaine for anesthesia relate to administration technique (resulting in systemic exposure) or pharmacological effects of anesthesia, and allergic reactions only rarely occur.[28] Systemic exposure to excessive quantities of lidocaine mainly result in central nervous system (CNS) and cardiovascular effects – CNS effects usually occur at lower blood plasma concentrations and additional cardiovascular effects present at higher concentrations, though cardiovascular collapse may also occur with low concentrations. ADRs by system are:
CNS excitation: nervousness, agitation, anxiety, apprehension, tingling around the mouth (circumoral paraesthesia), headache, hyperesthesia, tremor, dizziness, pupillary changes, psychosis, euphoria, hallucinations, and seizures
CNS depression with increasingly heavier exposure: drowsiness, lethargy, slurred speech, hypoesthesia, confusion, disorientation, loss of consciousness, respiratory depression and apnoea.
Skin: itching, depigmentation, rash, urticaria, edema, angioedema, bruising, inflammation of the vein at the injection site, irritation of the skin when applied topically
ADRs associated with the use of intravenous lidocaine are similar to toxic effects from systemic exposure above. These are dose-related and more frequent at high infusion rates (≥3 mg/min). Common ADRs include: headache, dizziness, drowsiness, confusion, visual disturbances, tinnitus, tremor, and/or paraesthesia. Infrequent ADRs associated with the use of lidocaine include: hypotension, bradycardia, arrhythmias, cardiac arrest, muscle twitching, seizures, coma, and/or respiratory depression.[29]
It is generally safe to use lidocaine with vasoconstrictor such as adrenaline, including in regions such as the nose, ears, fingers, and toes.[30] While concerns of tissue death if used in these areas have been raised, evidence does not support these concerns.[30]
Interactions
Any drugs that are also ligands of CYP3A4 and CYP1A2 can potentially increase serum levels and potential for toxicity or decrease serum levels and the efficacy, depending on whether they induce or inhibit the enzymes, respectively. Drugs that may increase the chance of methemoglobinemia should also be considered carefully. Dronedarone and liposomalmorphine are both absolutely a contraindication, as they may increase the serum levels, but hundreds of other drugs require monitoring for interaction.[31]
Contraindications
Absolute contraindications for the use of lidocaine include:
Heart block, second or third degree (without pacemaker)
Intra-articular infusion (this is not an approved indication and can cause chondrolysis)
Porphyria, especially acute intermittent porphyria; lidocaine has been classified as porphyrogenic because of the hepatic enzymes it induces,[34] although clinical evidence suggests it is not.[35]Bupivacaine is a safe alternative in this case.
Impaired liver function – people with lowered hepatic function may have an adverse reaction with repeated administration of lidocaine because the drug is metabolized by the liver. Adverse reactions may include neurological symptoms (e.g. dizziness, nausea, muscle twitches, vomiting, or seizures).[36]
Overdosage
Overdoses of lidocaine may result from excessive administration by topical or parenteral routes, accidental oral ingestion of topical preparations by children (who are more susceptible to overdose), accidental intravenous (rather than subcutaneous, intrathecal, or paracervical) injection, or from prolonged use of subcutaneous infiltration anesthesia during cosmetic surgery.
Such overdoses have often led to severe toxicity or death in both children and adults. Lidocaine and its two major metabolites may be quantified in blood, plasma, or serum to confirm the diagnosis in potential poisoning victims or to assist forensic investigation in a case of fatal overdose.
Lidocaine is often given intravenously as an antiarrhythmic agent in critical cardiac-care situations.[37] Treatment with intravenous lipid emulsions (used for parenteral feeding) to reverse the effects of local anaesthetic toxicity is becoming more common.[38]
Lidocaine alters signal conduction in neurons by prolonging the inactivation of the fast voltage-gated Na+ channels in the neuronal cell membrane responsible for action potential propagation.[40] With sufficient blockage, the voltage-gated sodium channels will not open and an action potential will not be generated. Careful titration allows for a high degree of selectivity in the blockage of sensory neurons, whereas higher concentrations also affect other types of neurons.
The same principle applies for this drug’s actions in the heart. Blocking sodium channels in the conduction system, as well as the muscle cells of the heart, raises the depolarization threshold, making the heart less likely to initiate or conduct early action potentials that may cause an arrhythmia.[41]
Pharmacokinetics
When used as an injectable it typically begins working within four minutes and lasts for half an hour to three hours.[8][9] Lidocaine is about 95% metabolized (dealkylated) in the liver mainly by CYP3A4 to the pharmacologically active metabolitesmonoethylglycinexylidide (MEGX) and then subsequently to the inactive glycine xylidide. MEGX has a longer half-life than lidocaine, but also is a less potent sodium channel blocker.[42] The volume of distribution is 1.1 L/kg to 2.1 L/kg, but congestive heart failure can decrease it. About 60% to 80% circulates bound to the protein alpha1 acid glycoprotein. The oral bioavailability is 35% and the topical bioavailability is 3%.
The elimination half-life of lidocaine is biphasic and around 90 min to 120 min in most patients. This may be prolonged in patients with hepatic impairment (average 343 min) or congestive heart failure (average 136 min).[43] Lidocaine is excreted in the urine (90% as metabolites and 10% as unchanged drug).[44]
History
Lidocaine, the first aminoamide–type local anesthetic, was first synthesized under the name ‘xylocaine’ by Swedish chemist Nils Löfgren in 1943.[45][46][47] His colleague Bengt Lundqvist performed the first injection anesthesia experiments on himself.[45] It was first marketed in 1949.
Society and culture
Dosage forms
Lidocaine, usually in the form of its hydrochloride salt, is available in various forms including many topical formulations and solutions for injection or infusion.[48] It is also available as a transdermal patch, which is applied directly to the skin.
Lidocaine hydrochloride 2% epinephrine 1:80,000 solution for injection in a cartridge
Lidocaine is often added to cocaine as a diluent.[54][55] Cocaine and lidocaine both numb the gums when applied. This gives the user the impression of high-quality cocaine, when in actuality the user is receiving a diluted product.[56]
^ Jump up to:abc J. P. Nolan; P. J. F. Baskett (1997). “Analgesia and anaesthesia”. In David Skinner; Andrew Swain; Rodney Peyton; Colin Robertson (eds.). Cambridge Textbook of Accident and Emergency Medicine. Project co-ordinator, Fiona Whinster. Cambridge, UK: Cambridge University Press. p. 194. ISBN9780521433792. Archived from the original on 2017-09-08.
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06.
^ Hamilton, Richart (2015). Tarascon Pocket Pharmacopoeia 2015 Deluxe Lab-Coat Edition. Jones & Bartlett Learning. p. 22. ISBN9781284057560.
^ Cepeda MS, Tzortzopoulou A, Thackrey M, Hudcova J, Arora Gandhi P, Schumann R (December 2010). Tzortzopoulou A (ed.). “Adjusting the pH of lidocaine for reducing pain on injection”. The Cochrane Database of Systematic Reviews (12): CD006581. doi:10.1002/14651858.CD006581.pub2. PMID21154371.(Retracted, see doi:10.1002/14651858.cd006581.pub3. If this is an intentional citation to a retracted paper, please replace{{Retracted}} with {{Retracted|intentional=yes}}.)
^ Sinha S, Schreiner AJ, Biernaskie J, Nickerson D, Gabriel VA (November 2017). “Treating pain on skin graft donor sites: Review and clinical recommendations”. The Journal of Trauma and Acute Care Surgery. 83 (5): 954–964. doi:10.1097/TA.0000000000001615. PMID28598907. S2CID44520644.
^ Biller JA (2007). “Airway obstruction, bronchospasm, and cough”. In Berger AM, Shuster JL, Von Roenn JH (eds.). Principles and practice of palliative care and supportive oncology. Hagerstwon, MD: Lippincott Williams & Wilkins. pp. 297–307. ISBN978-0-7817-9595-1. Inhaled lidocaine is used to suppress cough during bronchoscopy. Animal studies and a few human studies suggest that lidocaine has an antitussive effect…
^ Birsa LM, Verity PG, Lee RF (May 2010). “Evaluation of the effects of various chemicals on discharge of and pain caused by jellyfish nematocysts”. Comp. Biochem. Physiol. C. 151 (4): 426–30. doi:10.1016/j.cbpc.2010.01.007. PMID20116454.
^ Morabito R, Marino A, Dossena S, La Spada G (Jun 2014). “Nematocyst discharge in Pelagia noctiluca (Cnidaria, Scyphozoa) oral arms can be affected by lidocaine, ethanol, ammonia and acetic acid”. Toxicon. 83: 52–8. doi:10.1016/j.toxicon.2014.03.002. PMID24637105.
^ James G. Adams (2012). “32”. Emergency Medicine: Clinical Essentials. Elsevier Health Sciences. ISBN9781455733941. Archived from the original on 2017-09-08.
^ Jump up to:ab Nielsen LJ, Lumholt P, Hölmich LR (October 2014). “[Local anaesthesia with vasoconstrictor is safe to use in areas with end-arteries in fingers, toes, noses and ears]”. Ugeskrift for Laeger. 176(44). PMID25354008.
^“Lidocaine – N01BB02”. Drug porphyrinogenicity monograph. The Norwegian Porphyria Centre and the Swedish Porphyria Centre. Archived from the original on 2014-04-20. strong clinical evidence points to lidocaine as probably not porphyrinogenic
^ Khan, M. Gabriel (2007). Cardiac Drug Therapy (7th ed.). Totowa, NJ: Humana Press. ISBN9781597452380.
^ Baselt R (2008). Disposition of Toxic Drugs and Chemicals in Man(8th ed.). Foster City, CA: Biomedical Publications. pp. 840–4. ISBN978-0-9626523-7-0.
^ Picard J, Ward SC, Zumpe R, Meek T, Barlow J, Harrop-Griffiths W (February 2009). “Guidelines and the adoption of ‘lipid rescue’ therapy for local anaesthetic toxicity”. Anaesthesia. 64 (2): 122–5. doi:10.1111/j.1365-2044.2008.05816.x. PMID19143686. S2CID25581037.
^ Carterall, William A. (2001). “Molecular mechanisms of gating and drug block of sodium channels”. Sodium Channels and Neuronal Hyperexcitability. Novartis Foundation Symposia. 241. pp. 206–225. doi:10.1002/0470846682.ch14. ISBN9780470846681.
^ Jump up to:ab Löfgren N (1948). Studies on local anesthetics: Xylocaine: a new synthetic drug (Inaugural dissertation). Stockholm, Sweden: Ivar Heggstroms. OCLC646046738.[page needed]
^ Löfgren N, Lundqvist B (1946). “Studies on local anaesthetics II”. Svensk Kemisk Tidskrift. 58: 206–17.
^ Pupka A, Sikora J, Mauricz J, Cios D, Płonek T (2009). “[The usage of synthol in the body building]”. Polimery W Medycynie. 39(1): 63–5. PMID19580174.
^ Bernardo NP; Siqueira MEPB; De Paiva MJN; Maia PP (2003). “Caffeine and other adulterants in seizures of street cocaine in Brazil”. International Journal of Drug Policy. 14 (4): 331–4. doi:10.1016/S0955-3959(03)00083-5.
US patent 2441498, Nils Magnus Loefgren & Bengt Josef Lundqvist, “Alkyl glycinanilides”, published 1948-05-11, issued 1948-05-11, assigned to ASTRA APOTEKARNES KEM FAB

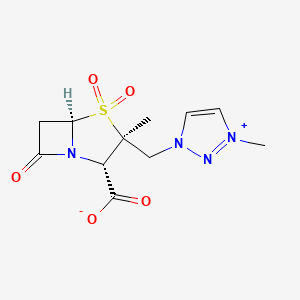













 by Alkermes Inc.
by Alkermes Inc.




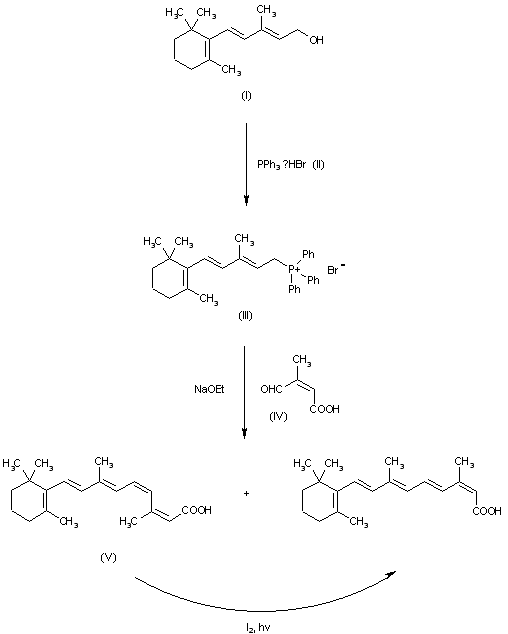
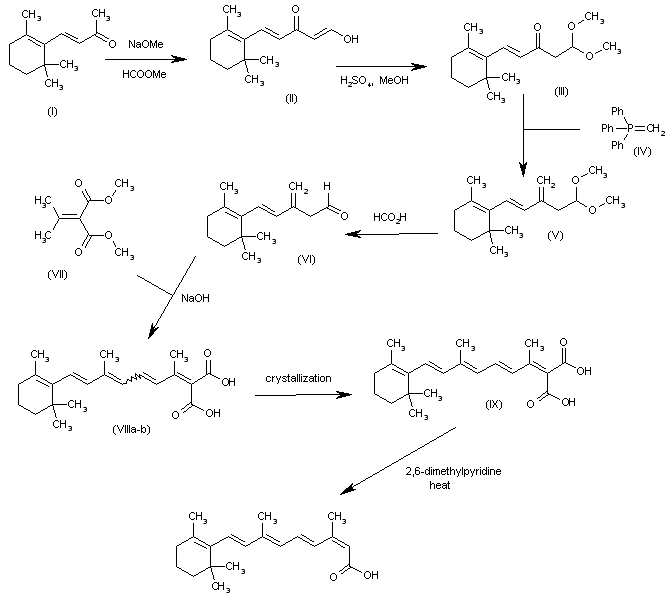














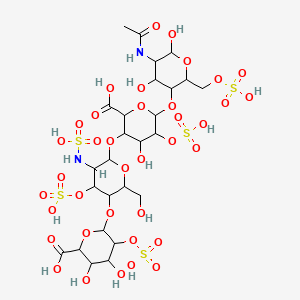



























































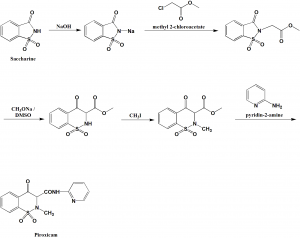




 Susanta Samajdar, Ph.D., Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited
Susanta Samajdar, Ph.D., Director, Medicinal Chemistry, Aurigene Discovery Technologies Limited






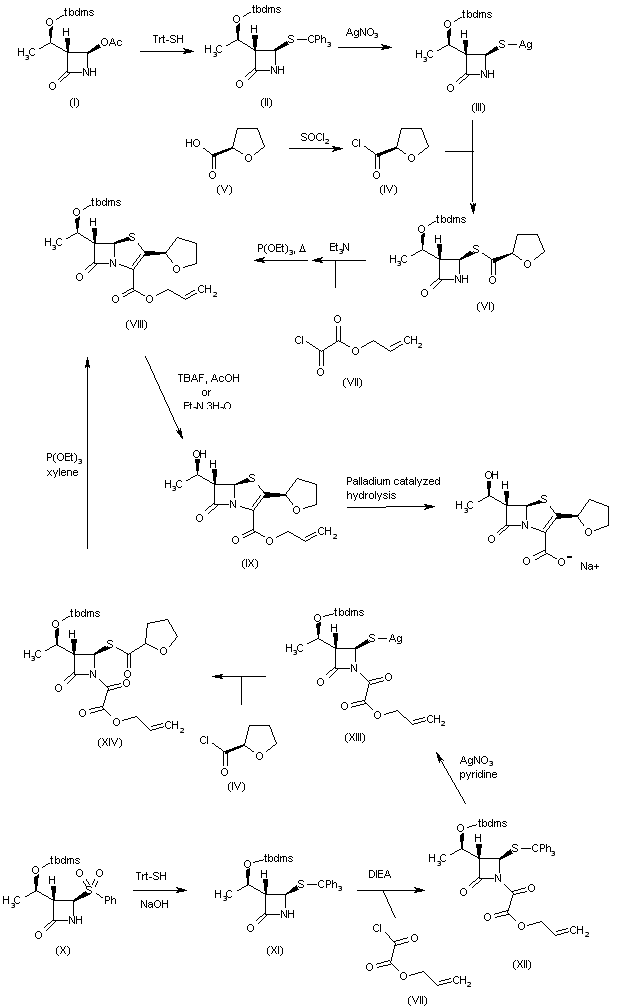
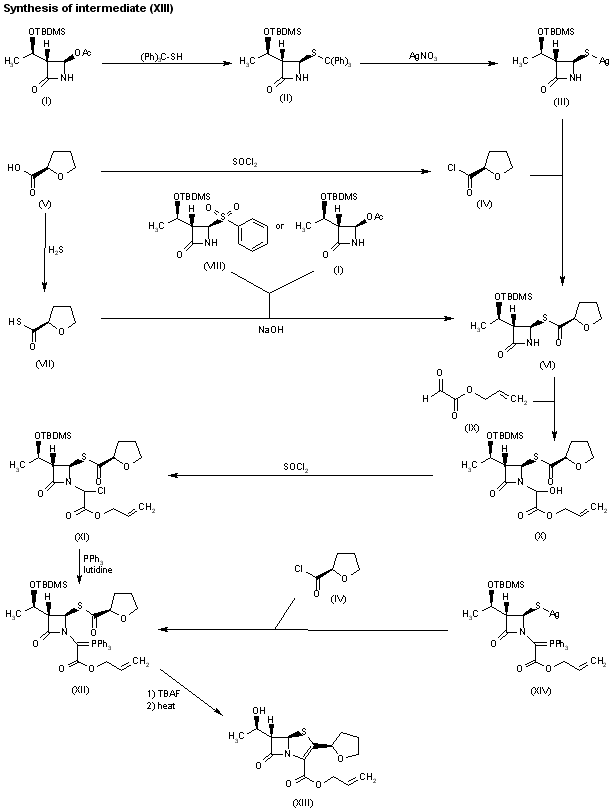
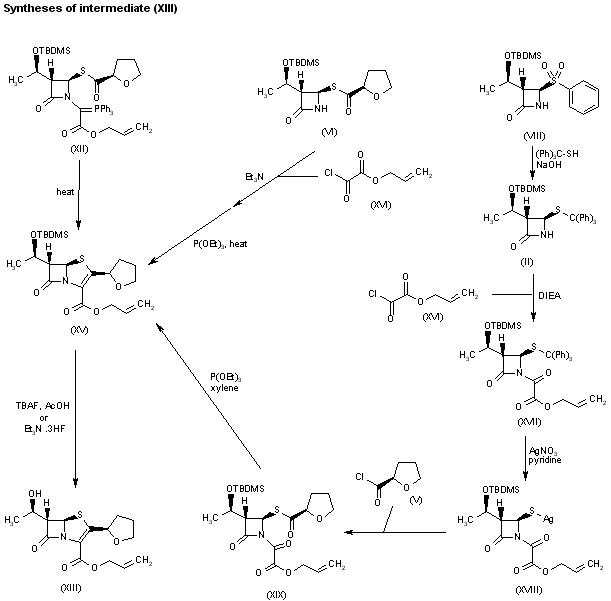
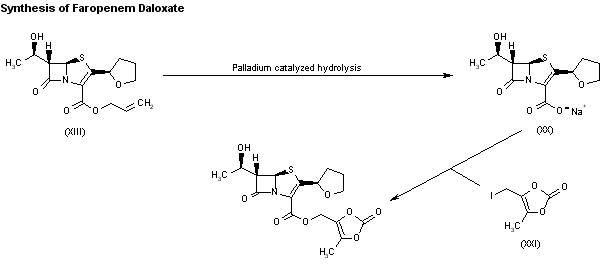

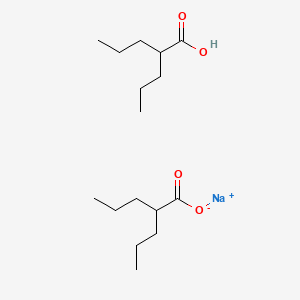









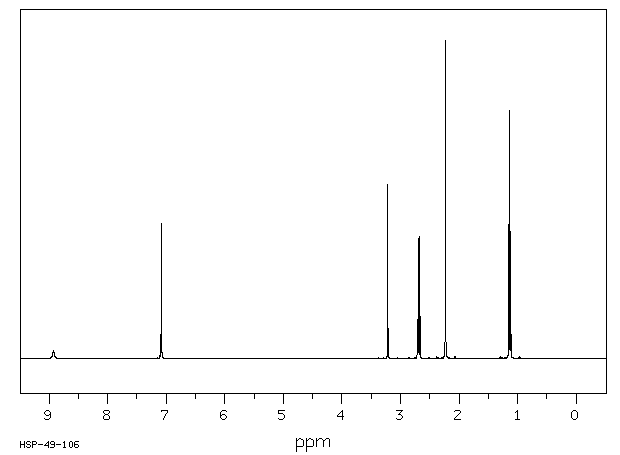
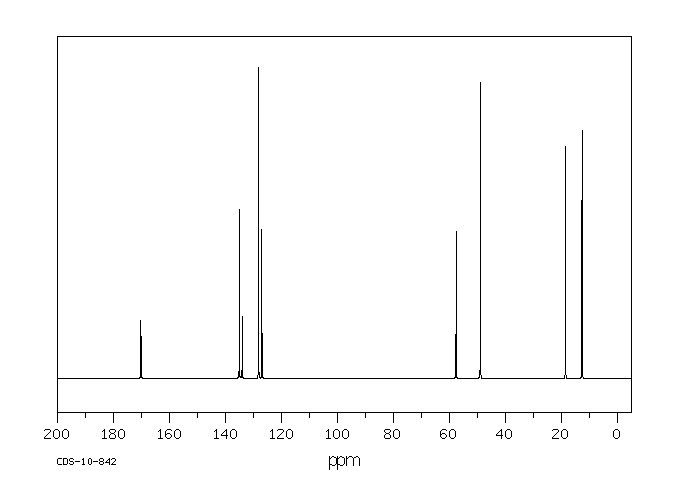
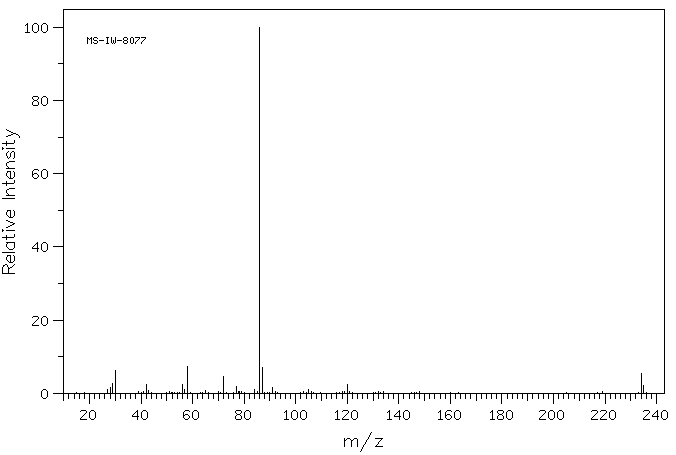
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)
.jpg)