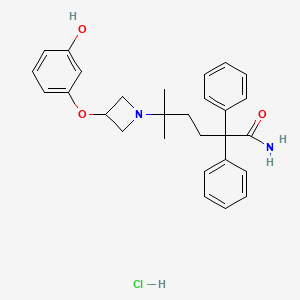
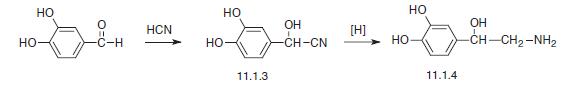
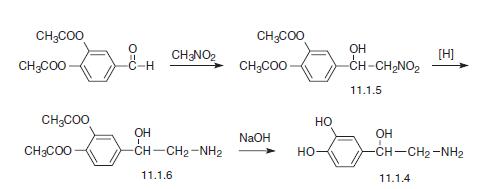
Certain glycopyrronium salts and related compounds, as well as processes for making and methods of using these glycopyrronium salts and related compounds, are known. See, for example, US Patent No. 8,558,008, which issued to assignee Dermira, Inc. See also, for example, US Patent No. 2,956,062, which issued to assignee Robins Co Inc. A H. See also, for example, International Patent Application Publication Nos. WO 98/00132 Al and WO 2009/00109A1, both of which list applicant Sepracor, Inc., as well as US Patent Nos. 6,063,808 and 6,204,285, both of which issued to assignee Sepracor, Inc. Certain methods of treating hyperhidrosis using glycopyrronium salts and related compounds are known. See, for example GB 1,080,960. Certain forms of applying glycopyrrolate compounds to a subject are known. See, for example US Patent Nos. 6,433,003 and 8,618,160, both of which issued to assignee Rose U; also US Patent Nos. 7,060,289; 8,252,316; and 8,679,524, which issued to PurePharm, Inc.
[0004] One glycopyrronium salt which is useful in certain medical applications is the following compound:
[0005] As illustrated above, the absolute configuration at the three asymmetric chiral positions is 2R3’R1’RS. This means that the carbon indicated with the number, 2, has the stereochemical R configuration. The carbon indicated with the number, 3′, also has the stereochemical R configuration. The quatemary ammonium nitrogen atom, indicated with a positive charge, may have either the R or the S stereochemical configuration. As drawn, the compound above is a mixture of two diastereoisomers.
[0006] Certain processes for making glycopyrronium salts are known. However, these processes are not as safe, efficient, stereospecific, or stereoselective as the new processes disclosed herein, for example with respect to large-scale manufacturing processes. Certain publications show that higher anticholinergic activity is attributed to the 2R3’R configuration. However, to date, processes for making the 2R3’R isomers, as well as the 2R3’R1’R isomers are low yielding, involve too many reaction steps to be economically feasible, use toxic materials, and/or are not sufficiently stereospecific or stereoselective with respect to the products formed.
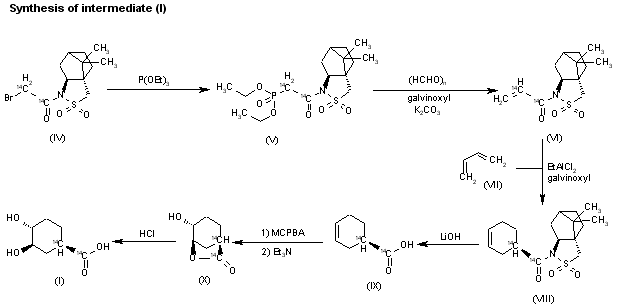
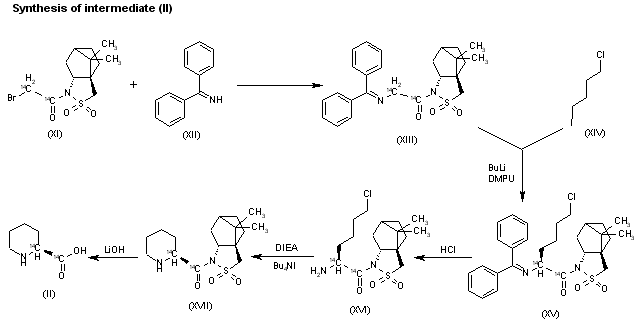
EXAMPLE 2
[0179] The below synthetic description refers to the numbered compounds illustrated in FIG. 2. Numbers which refer to these compounds in FIG. 2 are bolded and underlined in this Example.
[0180] Synthesis of R(-)-Cyclopentylmandelic acid (4)
[0181] R(-)-cyclopentylmandelic acid (compound 4) can be synthesized starting with
R(-)-mandelic acid (compound 1) according to Example 1.
[0182] Step 1 : Making Compound 2.
[0183] R(-)-mandelic acid (1) was suspended in hexane and mixed with pivaldehyde and a catalytic amount of trifluoromethanesulfonic acid at room temperature to form a mixture. The mixture was warmed to 36 °C and then allowed to react for about 5 hours. The mixture was then cooled to room temperature and treated with 8% aqueous sodium bicarbonate. The aqueous layer was removed and the organic layer dried over anhydrous sodium sulfate. After filtration and removal of the solvent under vacuum, the crude product was recrystallized to give (5R)-2-(tert-butyl)-5-phenyl-l,3-dioxolan-4-one (compound 2) in 88% yield (per S-enantiomer yield).
[0184] Step 2: Making Compound 3.
[0185] Compound 2 was reacted with lithium hexamethyl disilazide (LiHMDS) in hexane at -78 °C under stirring for one hour. Next, cyclopentyl bromide was added to the reaction mixture including compound 2 and LiHMDS . The reaction was kept cool for about four (4) hours and then slowly warmed to room temperature and allowed to react for at least twelve (12) more hours. The resulting mixture was then treated with 10% aqueous ammonium chloride. The aqueous layer was discarded and the organic layer dried over anhydrous sodium sulfate. The solvent was removed under vacuum and the residue recrystallized from hexane to give pure product (5R)-2-(tert-butyl)-5-cyclopentyl-5-phenyl- l,3-dioxolan-4-one (3) in 63% yield (per S-enantiomer yield).
[0186] Step 3: Making Compound 4.
[0187] R(-)-cyclopentylmandelic acid (compound 4) was prepared by providing compound 3 in aqueous methanolic potassium hydroxide at 65 °C for four hours. After cooling this mixture to room temperature and removing the methanol under vacuum, the aqueous solution was acidified with aqueous hydrochloric acid. The aqueous solution was then extracted twice with ethyl acetate and the organic phase dried with anhydrous sodium sulfate. After removing the solvent and performing a recrystallization, pure R(-)- cyclopentylmandelic acid (compound 4) was obtained in 62% yield (based on S-enantiomer yield).
[0188] Next, a racemic mixture of l -methyl-3-pyrridinol (20) was provided:
[0189] Synthesis of 2R3 ‘R-glycopyrrolate base (8)
[0190] Step 4: Making Compound 8.
[0191] Enantiomerically pure R(-)-cyclopentylmandelic acid (4) was coupled to racemic l-methyl-3-pyrridinol (20) using 1, 1 -carbonyldiimideazole (CDI) activated esterification to make an enantiomerically pure mixture of the following erythro- and threo- glycopyrrolate bases (compounds 8 and 21, respectively):
[0192] The 2R3’R-glycopyrrolate base (compound 8) was then resolved using the 5- nitroisophthalate salt procedure in Finnish Patent 49713, to provide enantiomerically pure 2R3 Έ. {erythro) as well as pure 2R3 ‘S {threo). In this example, the 2R3 ‘S {threo) was discarded. The 2R3 Έ. {erythro) was separated as stereomerically pure compound 8.
[0193] Step 6: Making Compound 9.
[0194] The glycopyrrolate base, compound 8, was treated in dry acetonitrile with methyl bromoacetate at room temperature under stirring for three (3) hours. The crude product was dissolved in a small volume of methylene chloride and poured into dry ethyl ether to obtain a precipitate. This procedure was repeated three times to provide (3R)-3-((R)- 2-cyclopentyl-2-hydroxy-2-phenylacetoxy)-l -(2-ethoxy-2-oxoethyl)-l-methylpyrrolidin-l – ium bromide, also known as 3′(R)-[R-Cyclopentylphenylhydroxyacetoy]- -ethyl- l ‘methoxycarbonylpyrrolidinium bromide (compound 9) in 89% yield. Compound 9 included the following stereoisomers:
Prevention and Treatment of Thromboembolic Disorders
Milvexian, also known as BMS-986177, is a blood coagulation factor XIa inhibitor.Bristol-Myers Squibb , in collaboration with Janssen , is developing milvexian (BMS-986177, JNJ-70033093; JNJ-3093), an antithrombotic factor XIa (FXIa) inhibitor, for the oral prevention and treatment of thrombosis.
Example 1. Preparation of (9i?,135)-13-{4-[5-chloro-2-(4-chloro-lH-l,2,3-triazol-l-yl)phenyl]-6-oxo- 1 ,6-dihydropyrimidin- 1 -yl} -3-(difluoromethyl)-9-methyl-3,4,7, 15-tetraazatricyclo[ 12.3.1.02‘6] -8-one trifluoroacetate
1A. Preparation of l-(difluoromethyl)-4-nitro-lH-pyrazole
CS2CO3 (14.41 g, 44.2 mmol) was suspended in a solution of 4-nitro-lH-pyrazole (5.00 g, 44.2 mmol) and DMF (40 mL). After heating to 120 °C for 5 min, solid sodium 2-chloro-2,2-difluoroacetate (13.48 g, 88 mmol) was added in 10 equal portions over 20 min. The reaction was complete after 10 min of additional heating. The mixture was added to a separatory funnel containing 100 mL water and extracted with Et20 (2 x 50 mL). The combined organic layers were concentrated. Purification by normal-phase chromatography eluting with a gradient of hexanes/EtOAc yielded l-(difluoromethyl)-4-nitro-lH-pyrazole (6.99 g, 42.9 mmol, 97% yield) as a clear, colorless oil. 1H NMR (500MHz, CDCI3) δ 8.58 (s, 1H), 8.22 (s, 1H), 7.39 – 7.05 (t, J= 60 Hz, 1H).
IB. Preparation of (S)-tert-butyl (l-(4-(l-(difluoromethyl)-4-nitro-lH-pyrazol-5-yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate
To a N2 flushed, 500 mL RBF was added {S)-tert-bvXy\ (l-(4-chloropyridin-2-yl)but-3-en-l-yl)carbamate, prepared as described in Example 3, (10 g, 35.4 mmol), 1-(difluoromethyl)-4-nitro-lH-pyrazol (6.34 g, 38.9 mmol) and dioxane (100 mL). The solution was bubbled with N2 for 5 min. Then Pd(OAc)2 (0.40 g, 1.7 mmol),
di(adamantan-l-yl)(butyl)phosphine (1.27 g, 3.5 mmol), K2CO3 (14.7 g, 106 mmol) and PvOH (1.08 g, 10.61 mmol) were added. The reaction mixture was bubbled with N2 for 5 min then the reaction mixture was heated to 100 °C for 3 h. After this time, the solution was cooled to rt and water (200 mL) was added. The reaction mixture was then extracted with EtOAc (2 x 200 mL). The combined organic extracts were washed with water (200 mL), brine (200 mL), dried over Na2S04, filtered and concentrated in vacuo. Purification by normal phase chromatography eluting with a gradient of hexanes/EtOAc afforded (S)-tert-butyl ( 1 -(4-( 1 -(difluoromethyl)-4-nitro- lH-pyrazol-5 -yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate (12.91 g, 31.5 mmol, 89% yield) as a slightly yellow oil. MS(ESI) m/z: 410.4 [M+H]+. 1H NMR (400MHz, CDC13) δ 8.80 (dd, J=5.1, 0.7 Hz, 1H), 8.36 (s, 1H), 7.34 (s, 1H), 7.31 (dd, J=5.1, 1.5 Hz, 1H), 7.27 – 6.91 (t, J=58 Hz, 1H), 5.79 – 5.63 (m, 1H), 5.16 – 5.03 (m, 2H), 4.92 (d, J=5.9 Hz, 1H), 2.67 (t, J=6.4 Hz, 2H), 1.46 (br. s., 9H).
1C. Preparation of (l-(4-(4-amino-l -(difluoromethyl)- lH-pyrazol-5-yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate
To a 100 mL, 3-necked RBF was added a solution of (S)-tert-butyl (l-(4-(l-(difluoromethyl)-4-nitro-lH-pyrazol-5-yl)pyridin-2-yl)but-3-en-l-yl)carbamate (0.78 g, 1.90 mmol) in MeOH (12 mL) and a solution of NH4C1 (1.02 g, 19 mmol) in water (3 mL). To the solution was added Fe (0.53 g, 9.49 mmol). The reaction mixture was heated to 65 °C for 3 h. Water (50 mL) was added. After cooling to rt, the mixture was filtered through a CELITE® pad and rinsed with MeOH (200 mL). The filtrate was concentrated in vacuo. The residue was partitioned between EtOAC (100 mL) and water (100 mL). The organic phase was separated, washed with water (100 mL), brine (100 mL), dried over Na2S04, filtered and concentrated in vacuo. Purification by normal phase chromatography eluting with a gradient of DCM/MeOH yielded (S)-tert-butyl (l-(4-(4-amino- 1 -(difluoromethyl)- lH-pyrazol-5 -yl)pyridin-2-yl)but-3 -en- 1 -yl)carbamate (0.585 g, 1.54 mmol, 81% yield) as an oil. MS(ESI) m/z: 380.1 [M+H]+. 1H NMR (400MHz,
ID. Preparation of tert-butyl ((5)-l-(4-(l-(difiuoromethyl)-4-((i?)-2-methylbut-3-enamido)- lH-pyrazol-5-yl)pyridin-2-yl)but-3-en- 1 -yl)carbamate
To a N2 flushed, 3 -necked, 250 mL RBF was added a solution of {S)-tert-bvXy\ (1-(4-(4-amino-l-(difluoromethyl)-lH-pyrazol-5-yl)pyridin-2-yl)but-3-en-l-yl)carbamate (5 g, 13.18 mmol) and EtOAc (50 ml). The solution was cooled to -10 °C and (R)-2-methylbut-3-enoic acid, as prepared in Example 2, (1.72 g, 17.13 mmol), pyridine (4.26 ml, 52.7 mmol). and T3P® (23.54 ml, 39.5 mmol) were added. The cooling bath was removed and the solution was allowed to warm to rt and then stir over a period of 20 h. Water (30 mL) and EtOAc (30 mL) were added and the mixture was stirred for 30 min. The organic phase was separated and the aqueous layer was extracted with EtOAc (30 mL). The combined organic extracts were washed with brine (50 mL), dried over
Na2SC”4, filtered and concentrated in vacuo. Purification by normal phase
IE. Preparation of tert-butyl N-[(9i?,10E,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,10,14,16-hexaen-13-yl] carbamate
To a N2 flushed, 2 L, 3 -necked, RBF was added a solution of tert-butyl ((S)-l-(4-(1 -(difluoromethyl)-4-((i?)-2-methylbut-3 -enamido)- lH-pyrazol-5 -yl)pyridin-2-yl)but-3 -en-l-yl)carbamate (3 g, 6.50 mmol) in EtOAc (1300 ml). The solution was sparged with argon for 15 min. Grubbs II (1.38 g, 1.63 mmol) was added in one portion. The reaction mixture was heated to reflux for 24 h. After cooling to rt, the solvent was removed and the residue was purified by normal phase chromatography eluting with a gradient of DCM/MeOH to yield tert-butyl N-[(9R, 10E, 135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,10,14,16-hexaen-13-yl]carbamate (2.13 g, 4.91 mmol, 76% yield) as a tan solid. MS(ESI) m/z: 434.4 [M+H]+. 1H NMR (400MHz, CDC13) δ 8.71 (d, J=5.1 Hz, 1H), 7.78 (s, 1H), 7.44 – 7.40 (m, 1H), 7.36 (br. s., 1H), 7.27 (t, J=58 Hz, 1H), 6.87 (s, 1H), 6.49 – 6.39 (m, 1H), 5.78 (s, 1H), 4.80 (br. s., 2H), 3.18 – 3.08 (m, 1H), 3.08 – 2.98 (m, 1H), 2.06 – 1.93 (m, 1H), 1.51 (s, 9H), 1.19 (d, J=6.6 Hz, 3H).
IF. Preparation of tert-butyl N-[(9i?,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-13-yl]carbamate
Pd/C (0.60 g, 0.570 mmol) was added to a 250 mL Parr hydrogenation flask containing a solution of tert-butyl N-[(9i?,10E,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,10,14,16-hexaen-13-yljcarbamate (2.46 g, 5.68 mmol) in EtOH (100 mL). The flask was purged with N2 and pressurized to 55 psi of H2 allowed to stir for 18 h. The reaction was filtered through CELITE® and concentrated to yield tert-butyl N-[(9i?,135)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-13-yl]carbamate (2.17 g, 88% yield) as a tan solid. MS(ESI) m/z: 436.3 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 9.32 (s, 1H), 8.71 (d, J=5.0 Hz, 1H), 7.96 (t, J=58 Hz, 1H), 7.43 (s, 1H), 7.32 (d, J=4.8 Hz, 1H), 7.22 (d, J=7.3 Hz, 1H), 4.66 (d, J=8.3 Hz, 1H), 2.62 (br. s., 1H), 1.88 (d, J=12.8 Hz, 1H), 1.77 – 1.59 (m, 2H), 1.42 – 1.28 (m, 9H), 1.15 (d, J=18.2 Hz, 2H), 0.83 (d, J=7.0 Hz, 3H).
I G. Preparation of (9R, 13S)-l 3-amino-3-(difiuoromethyl)-9-methyl-3,4,7, 15-tetraazatricyclo[ 12.3.1.02‘6]octadeca- 1(18),2(6),4, 14,16-pentaen-8-one
4 N HC1 in dioxane (3.88 mL, 15.5 mmol) was added to a solution of tert-butyl N-[(9R, 13S)-3-(difluoromethyl)-9-methyl-8-oxo-3,4,7, 15-tetraazatricyclo[12.3.1.02‘6] octadeca-l(18),2(6),4,14,16-pentaen-13-yl]carbamate (2.25 g, 5.2 mmol) in MeOH (10 mL). The reaction was allowed to stir at rt for 2 h. The reaction was cooled in an ice bath, and 7 N NH3 in MeOH (13.3 mL, 93.0 mmol) was added. After 5 min, the reaction was diluted with CH2C12 (80 mL) and the solid that formed was filtered. The filtrate was concentrated to yield (9i?,135)-13-amino-3-(difluoromethyl)-9-methyl-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-8-one (1.3 g, 3.88 mmol, 75% yield). MS(ESI) m/z: 336.3 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 9.33 (s, 1H), 8.71 (d, J=5.0 Hz, 1H), 7.94 (t, J=58 Hz, 1H), 7.85 (s, 1H), 7.40 (s, 1H), 7.32 (d, J=5.0 Hz, 1H), 4.01 (dd, J=10.2, 5.1 Hz, 1H), 2.63 – 2.53 (m, 1H), 1.90 – 1.69 (m, 2H), 1.53 -1.36 (m, 2H), 1.16 – 1.00 (m, 1H), 0.85 (d, J=7.0 Hz, 3H).
To a 100 mL flask containing a white suspension of 6-(5-chloro-2-(4-chloro-lH-l,2,3-triazol-l-yl)phenyl)pyrimidin-4-ol (0.83 g, 2.7 mmol), as prepared in Example 4 in ACN (36 mL) was added HATU (1.12 g, 3.0 mmol) and DBU (0.53 mL, 3.5 mmol). The resulting clear, yellow solution was stirred at rt. After 5 min, (9i?,135)-13-amino-3-(difluoromethyl)-9-methyl-3,4,7,15-tetraazatricyclo[12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-8-one (0.9 g, 2.68 mmol) was added and the resulting suspension was stirred at rt for 3 h. The reaction was then concentrated and purified by normal phase silica gel chromatography, eluting with a gradient of 0% to 100% EtOAc in hexanes to yield (9i?,135)-13-{4-[5-chloro-2-(4-chloro-lH-l,2,3-triazol-l-yl)phenyl]-6-oxo- 1 ,6-dihydropyrimidin- 1 -yl} -3-(difluoromethyl)-9-methyl-3 ,4,7, 15-tetraazatricyclo [12.3.1.02‘6]octadeca-l(18),2(6),4,14,16-pentaen-8-one (0.87 g, 50% yield) as a white solid. MS(ESI) m/z: 626.2 [M+H]+. 1H NMR (500MHz, CD3OD) δ 8.91 – 8.83 (m, 1H), 8.78 – 8.71 (m, 1H), 8.33 (s, 1H), 7.88 (d, J=2.5 Hz, 1H), 7.74 (s, 2H), 7.69 – 7.67 (m, 1H), 7.65 (s, 1H), 7.63 (t, J=58 Hz, 1H), 7.52 – 7.50 (m, 1H), 6.36 (d, J=0.8 Hz, 1H),
Spinosyn A: The chemical name is: 1H-as-Indaceno[3,2- d]oxacyclododecin-7,a5-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-alphaL-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-metyl-, (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-
Spinosyn D: The chemical name is: 1H-as-Indaceno[3,2- d]oxacyclododecin-7,15-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-alphaL-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-4,14-dimetyl-, (2S,3aSR,5aS,5bS,9S,13S,14R,16aS,16bS)-
168316-95-8
Molecular FormulaC83H132N2O20
Average mass1477.938 Da
Comfortis
Conserve
EC 434-300-1
Natroba
NaturaLyte
Spinosad
Tracer
Tracer Naturalyte
UNII-XPA88EAP6V
XDE 105
Natroba (Spinosad) Suspension 0.9% ParaPro Pharma
New Drug Application (NDA): 022408 appr 01/18/2011
spinosad, is a new molecular entity, and a fermentation product produced by the actinomycete, Saccharopolyspora spinosa. Spinosad contains two components, spinosyn A and D. T
Figure 1. Structure of spinosyn A and DTitle: SpinosynsCAS Registry Number: 131929-60-7Literature References: Class of fermentation derived 12 membered macrocyclic lactones in a unique tetracyclic ring. At least 20 spinosyns have been isolated from Saccharopolyspora spinosa; variations in the two sugars account for most of the structural and insecticidal activity differences. Isolation and biological activity: L. D. Boeck et al.,EP375316 (1990 to Lilly); eidem,US5496931 (1996 to DowElanco); and structure determn: H. A. Kirst et al.,Tetrahedron Lett.32, 4839 (1991). Soil degradation: K. A. Hale, D. E. Portwood, J. Environ. Sci. HealthB31, 477 (1996). HPLC determn in vegetables: L.-T. Yeh et al.,J. Agric. Food Chem.45, 1746 (1997); in soil and water: S. D. West, ibid. 3107. Uptake and metabolism in larvae: T. C. Sparks et al.,Proc. Beltwide Cotton Conf.2, 1259 (1997). Mode of action study: V. L. Salgado et al.,Pestic. Biochem. Physiol.60, 103 (1998). Review of physical and biological properties: C. V. DeAmicis et al.,ACS Symp. Ser.658, 144-154 (1997). Review: G. D. Crouse, T. C. Sparks, Rev. Toxicol.2, 133-146 (1998). Derivative Type: Spinosyn Acas 131929-60-7CAS Name: (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-2-[(6-Deoxy-2,3,4-tri-O-methyl-a-L-mannopyranosyl)oxy]-13-[[(2R,5S,6R)-5-(dimethylamino)tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-methyl-1H-as-indaceno[3,2-d]oxacyclododecin-7,15-dioneAdditional Names: lepicidin AManufacturers’ Codes: A-83543A; LY-232105Molecular Formula: C41H65NO10Molecular Weight: 731.96Percent Composition: C 67.28%, H 8.95%, N 1.91%, O 21.86%Literature References: Total synthesis: L. A. Paquette et al.,J. Am. Chem. Soc.120, 2553 (1998).Properties: White, odorless crystalline solid, mp 118°. pKa 8.1. uv max (methanol): 243 nm (e 11000). [a]27436 -262.7° (methanol). Vapor pressure: 2.4 ´ 10-10. Soly in water (ppm): 290 (pH 5), 235 (pH 7), 16 (pH 9), distilled 20. Soly (w/v%): methanol 19, acetone 17, dichloromethane >50, hexane 0.45%. LD50 in rats (mg/kg): 3783-5000 orally (Crouse).Melting point: mp 118°pKa: pKa 8.1Optical Rotation: [a]27436 -262.7° (methanol)Absorption maximum: uv max (methanol): 243 nm (e 11000)Toxicity data: LD50 in rats (mg/kg): 3783-5000 orally (Crouse) Derivative Type: Spinosyn DCAS Registry Number: 131929-63-0Manufacturers’ Codes: A-83543DMolecular Formula: C42H67NO10Molecular Weight: 745.98Percent Composition: C 67.62%, H 9.05%, N 1.88%, O 21.45%Properties: Odorless, white crystalline solid. mp 169°. pKa 7.8. uv max (methanol): 243 nm (e 11000). [a]27436 -297.5° (methanol). Vapor pressure: 2.0 ´ 10-10. Soly in water (ppm): 28 (pH 5), 0.329 (pH 7), 0.04 (pH 9), distilled 1.3. Soly (w/v%): methanol 0.25, acetone 1.0, dichloromethane 45, hexane 0.07%.Melting point: mp 169°pKa: pKa 7.8Optical Rotation: [a]27436 -297.5° (methanol)Absorption maximum: uv max (methanol): 243 nm (e 11000) Derivative Type: SpinosadCAS Registry Number: 168316-95-8Manufacturers’ Codes: XDE-105; DE-105Trademarks: Conserve (Dow AgroSci.); Justice (Dow AgroSci.); Naturalyte (Dow AgroSci.); SpinTor (Dow AgroSci.); Success (Dow AgroSci.); Tracer (Dow AgroSci.)Literature References: Mixture of spinosyns A and D. Effect on beneficial insects: D. Murray, R. Lloyd, Australian Cottongrower18, 62 (1997).Properties: Light grey to white crystals (tech). LD50 in rats, mallard ducks, quail (mg/kg): >3600, >2000, >2000 orally (Crouse).Toxicity data: LD50 in rats, mallard ducks, quail (mg/kg): >3600, >2000, >2000 orally (Crouse) Use: Insecticide.(2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-13-{[(2R,5S,6R)-5-(Dimethylamino)-6-methyltetrahydro-2H-pyran-2-yl]oxy}-9-ethyl-14-methyl-7,15-dioxo-2,3,3a,5a,5b,6,7,9,10,11,12,13,14,15,16a,16b-hexadecahydro-1H ;-as-indaceno[3,2-d]oxacyclododecin-2-yl 6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranoside – (2S,3aR,5aS,5bS,9S,13S,14R,16aS,16bS)-13-{[(2R,5S,6R)-5-(dimethylamino)-6-methyltetrahydro-2H-pyran-2-yl]ox y}-9-ethyl-4,14-dimethyl-7,15-dioxo-2,3,3a,5 1H-as-Indaceno[3,2-d]oxacyclododecin-7,15-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)oxy]-13-[[(2R,5S,6R)-5-(dimethylamino)tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b ,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-methyl-, (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-, compd. with (2S,3aR,5aS,5bS,9S,13S,14R,16aS,16bS)-2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)o xy]-13-[[(2R,5S,6R)-5-(dimethylamino)tetrahySpinosad[USAN] [Wiki]168316-95-8 [RN]1H-as-Indaceno[3,2-d]oxacyclododecin-7,15-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-4,14-dimetyl-,(2S,3aSR,5aS,5bS,9S,13S,14R,16aS,16bS)-1H-as-Indaceno[3,2-d]oxacyclododecin-7,a5-dione, 2-[(6-deoxy-2,3,4-tri-O-methyl-α-L-mannopyranosyl)oxy]-13-[[2R,5S,6R)-5-(dimethylamino) tetrahydro-6-methyl-2H-pyran-2-yl]oxy]-9-ethyl-2,3,3a,5a,5b,6,9,10,11,12,13,14,16a,16b-tetradecahydro-14-metyl-, (2R,3aS,5aR,5bS,9S,13S,14R,16aS,16bR)-NAF-144Spinosad|spinosyn A and D (mixture)spinosyn A and D (mixture)
Spinosad is an insecticide based on chemical compounds found in the bacterial species Saccharopolyspora spinosa. The genus Saccharopolyspora was discovered in 1985 in isolates from crushed sugarcane. The bacteria produce yellowish-pink aerial hyphae, with bead-like chains of spores enclosed in a characteristic hairy sheath.[1] This genus is defined as aerobic, Gram-positive, nonacid-fast actinomycetes with fragmenting substrate mycelium. S. spinosa was isolated from soil collected inside a nonoperational sugar mill rum still in the Virgin Islands. Spinosad is a mixture of chemical compounds in the spinosyn family that has a generalized structure consisting of a unique tetracyclic ring system attached to an amino sugar (D-forosamine) and a neutral sugar (tri-Ο-methyl-L-rhamnose).[2] Spinosad is relatively nonpolar and not easily dissolved in water.[3]
Spinosad is a novel mode-of-action insecticide derived from a family of natural products obtained by fermentation of S. spinosa. Spinosyns occur in over 20 natural forms, and over 200 synthetic forms (spinosoids) have been produced in the lab.[4] Spinosad contains a mix of two spinosoids, spinosyn A, the major component, and spinosyn D (the minor component), in a roughly 17:3 ratio.[1
Mode of action
Spinosad is highly active, by both contact and ingestion, in numerous insect species.[5] Its overall protective effect varies with insect species and life stage. It affects certain species only in the adult stage, but can affect other species at more than one life stage. The species subject to very high rates of mortality as larvae, but not as adults, may gradually be controlled through sustained larval mortality.[5] The mode of action of spinosoid insecticides is by a neural mechanism.[6] The spinosyns and spinosoids have a novel mode of action, primarily targeting binding sites on nicotinic acetylcholine receptors (nAChRs) of the insect nervous system that are distinct from those at which other insecticides have their activity. Spinosoid binding leads to disruption of acetylcholine neurotransmission.[2] Spinosad also has secondary effects as a γ-amino-butyric acid (GABA) neurotransmitter agonist.[2] It kills insects by hyperexcitation of the insect nervous system.[2] Spinosad so far has proven not to cause cross-resistance to any other known insecticide.[7]
Use
Spinosad has been used around the world for the control of a variety of insect pests, including Lepidoptera, Diptera, Thysanoptera, Coleoptera, Orthoptera, and Hymenoptera, and many others.[8] It was first registered as a pesticide in the United States for use on crops in 1997.[8] Its labeled use rate is set at 1 ppm (1 mg a.i./kg of grain) and its maximum residue limit (MRL) or tolerance is set at 1.5 ppm. Spinosad’s widespread commercial launch was deferred, awaiting final MRL or tolerance approvals in a few remaining grain-importing countries. It is considered a natural product, thus is approved for use in organic agriculture by numerous nations.[5] Two other uses for spinosad are for pets and humans. Spinosad has recently been used in oral preparations (as Comfortis) to treat C. felis, the cat flea, in canines and felines; the optimal dose set for canines is reported to be 30 mg/kg.[2]
Spinosad is sold under the trade names, Comfortis, Trifexis, and Natroba.[9][10] Trifexis also includes milbemycin oxime. Comfortis and Trifexis brands treat adult fleas on pets; the latter also prevents heartworm disease. Natroba is sold for treatment of human head lice. Spinosad is also commonly used to kill thrips.[11][12][13]
Spinosyn A
Spinosyn A does not appear to interact directly with known insecticidal-relevant target sites, but rather acts via a novel mechanism.[6] Spinosyn A resembles a GABA antagonist and is comparable to the effect of avermectin on insect neurons.[4] Spinosyn A is highly active against neonate larvae of the tobacco budworm, Heliothis virescens, and is slightly more biologically active than spinosyn D. In general, spinosyns possessing a methyl group at C6 (spinosyn D-related analogs) tend to be more active and less affected by changes in the rest of the molecule.[7] Spinosyn A is slow to penetrate to the internal fluids of larvae; it is also poorly metabolized once it enters the insect.[7] The apparent lack of spinosyn A metabolism may contribute to its high level of activity, and may compensate for the slow rate of penetration.[7]
Safety and ecotoxicology
Spinosad has high efficacy, a broad insect pest spectrum, low mammalian toxicity, and a good environmental profile, a unique feature of the insecticide compared to others currently used for the protection of grain products.[5] It is regarded as natural product-based, and approved for use in organic agriculture by numerous national and international certifications.[8] Spinosad residues are highly stable on grains stored in bins, with protection ranging from 6 months to 2 years.[5][clarification needed] Ecotoxicology parameters have been reported for spinosad, and are:[14]
in rat (Rattus norvegicus Bergenhout, 1769), acute oral: LD50 >5000 mg/kg (nontoxic)
in rat (R. norvegicus), acute dermal: LD50 >2000 mg/kg (nontoxic)
in California quail (Callipepla californica Shaw, 1798), oral toxicity: LD50 >2000 mg/kg (nontoxic)
in Honeybee (Apis mellifera Linnaeus, 1758), LD50 = 0.0025 mg/bee (highly toxic if directly sprayed on and of dried residues).
Chronic exposure studies failed to induce tumor formation in rats and mice; mice given up to 51 mg/kg/day for 18 months resulted in no tumor formation.[15] Similarly, administration of 25 mg/kg/day to rats for 24 months did not result in tumor formation.[16]
Pleocidin compounds (spinosyns) is soil actinomycete thorn many armfuls of bacterium Saccharopolysporaspinosa of sugar secondary metabolites behind aerobic fermentation under developing medium.Pleocidin belongs to macrolides compound, it comprises one a plurality of chiral carbon tetracyclic ring systems (Macrolide tetracycle), big ring is gone up the 9-hydroxyl and is being linked two different hexa-atomic sugar respectively with the 17-hydroxyl, wherein that 17 connections is an aminosugar (Forosamine sugar), and that connect on the 9-position is a rhamnosyl (Rhamnose sugar).Tetracyclic ring system is by one 5,, 6,5-is suitable-and anti–anti–three-loop system condenses one 12 membered macrolide to be formed, and wherein contains an alpha, beta-unsaturated ketone and an independently two key.When 6 on ring is pleocidin A when being substituted by hydrogen, in mixture, account for 85-90%, when ring 6 bit substituents when connecing methyl, be pleocidin D then, in mixture, account for about 10-15%.Up to the present B, C, D, E, F, G, K, L, M, N, O, P, Q, R, S, T, U, more than 20 derivative such as V, W etc. have been found and have isolated it to comprise Spinosyn A.
The commercialization kind has pleocidin Spinosyns (mixture of pleocidin A and pleocidin D) at present, the s-generation pleocidin insecticides Spinetoram. latter is got through semisynthesis by the thick product pleocidin L of biological method preparation and the mixture of J, promptly by 5 of pleocidin J, 6 two key selective reductions, reach 3 ‘ O-ethylization of rhamnosyl and obtain its major ingredient, ethylizing by 3 ‘ O-of pleocidin L rhamnosyl obtains its minor consistuent.
The pleocidin compound can be controlled lepidopteran, Diptera and Thysanoptera insect effectively.It can prevent and treat the pest species of some blade of eating in a large number in Coleoptera and the Orthoptera well.Pleocidin has very high activity to lepidopterous larvaes such as Heliothis virescens, bollworm, beet armyworm, prodenia litura, cabbage looper, small cabbage moth and rice-stem borers, and they are suitable environmental protection, have interesting toxicology character.
U.S. Patent No. 5362634 discloses the derivative that natural pleocidin is replaced by methyl or ethyl on C-21, U.S. Patent application No.60/153513 has disclosed the natural butenyl pleocidin derivative that the 3-4 carbochain replaces on C-21.Pleocidin derivative (John Daeuble, ThomasC.Sparks, Peter Johnson, Paul R.Graupner, the Bioorganic ﹠amp that can prepare C-21 position different substituents by replacement(metathesis)reaction; Medicinal Chemistry17 (2009) 4197-4205).U.S. Patent No. 6001981A, WO 9700265A have openly opened the chemosynthesis of pleocidin compound and have modified, and comprise aminosugar and rhamnosyl and the big chemically modified that encircles in the structure.
Spinosyns (A83543) are produced by derivatives of Saccharopolyspora spinosa NRRL18395 including strains NRRL 18537, 18538, 18539, 18719, 18720, 18743 and 18823 and derivatives thereof. A more preferred nomenclature for spinosyns is to refer to the pseudoaglycones as spinosyn A 17-Psa, spinosyn D 17-Psa, etc., and to the reverse pseudoaglycones as spinosyn A 9-Psa, spinosyn D 9-Psa, etc. (see Kirst et al., 1991). The known members of this family have been referred to as factors or components, and each has been given an identifying letter designation. These compounds are hereinafter referred to as spinosyn A, B, etc. The spinosyn compounds are useful for the control of arachnids, nematodes and insects, in particular Lepidoptera and Diptera species, and they are quite environmentally friendly and have an appealing toxicological profile. [0004] U.S. Patent No. 5,362,634 and corresponding European Patent Application No. 375316 Al disclose spinosyns A, B, C, D, E, F, G, H, and J. WO 93/09126 discloses spinosyns L, M, N, Q, R, S, and T. WO 94/20518 and US 5,6704,486 disclose spinosyns K,
O, P, U, V, W, and Y, and derivatives thereof. A large number of synthetic modifications to spinosyn compounds have been made, as disclosed in U.S. Patent No. 6,001,981 and WO
^ Jump up to:abcde Qiao, Meihua; Daniel E. Snyder; Jeffery Meyer; Alan G. Zimmerman; Meihau Qiao; Sonya J. Gissendanner; Larry R. Cruthers; Robyn L. Slone; Davide R. Young (12 September 2007). “Preliminary Studies on the effectiveness of the novel pulicide, spinosad, for the treatment and control of fleas on dogs”. Veterinary Parasitology. 150 (4): 345–351. doi:10.1016/j.vetpar.2007.09.011. PMID17980490.
^ Crouse, Gary; Thomas C Sparks; Joseph Schoonover; James Gifford; James Dripps; Tim Brue; Larry L Larson; Joseph Garlich; Chris Hatton; Rober L Hill; Thomas V Worden; Jacek G Martynow (27 September 2000). “Recent advances in the chemistry of spinosyns”. Pest Manag Sci. 57 (2): 177–185. doi:10.1002/1526-4998(200102)57:2<177::AID-PS281>3.0.CO;2-Z. PMID11455648.
^ Jump up to:ab Watson, Gerald (31 May 2001). “Actions of Insecticidal Spinosyns on gama-Aminobutyric Acid Responses for Small-Diameter Cockroach Neurons”. Pesticide Biochemistry and Physiology. 71: 20–28. doi:10.1006/pest.2001.2559.
^ Jump up to:ab Orr, Nailah; Andrew J. Shaffner; Kimberly Richey; Gary D. Crouse (30 April 2009). “Novel mode of action of spinosad: Receptor binding studies demonstrating lack of interaction with known insecticidal target sites”. Pesticide Biochemistry and Physiology. 95: 1–5. doi:10.1016/j.pestbp.2009.04.009.
^ Jump up to:abcd Sparks, Thomas; Gary D crouse; Gregory Durst (30 March 2001). “Natural products as insecticides: the biology, biochemistry and quantitative structure-activity relationships of spinosyns and spinosoids”. Pest Manag Sci. 57 (10): 896–905. doi:10.1002/ps.358. PMID11695182.
Spinosad is produced directly from the fermentation of a strain of Saccharo-polyspora spinosa. Production strains of S. spinosa have been selected for increased titers of spinosyns A and D, however, no genetic engineering techniques have been used in this process and no genetically-modified organisms are used in the production process. After fermentation, the spinosyn A and D mixture is extracted from the fermentation broth, precipitated and dried to create technical spinosad, which is then formulated into end-use products. Spinosad technical material is also produced under pharmaceutical manufacturing guidelines to be used as a flea control agent in companion animals.
5.9.2 Production of Spinetoram
Production of spinetoram begins with the fermentation of a mutant strain of Saccharopolyspora spinosa that produces primarily spinosyns J and L, rather than spinosyns A and D. This strain was generated through mutagenesis of S. spinosa. However, like the spinosad-producing strains, no genetic engineering techniques were used in this process and no genetically-modified organisms are used in the production process. After fermentation, the spinosyn J and L mixture is extracted from the fermentation broth and precipitated in preparation for the two chemical synthesis steps required to produce spinetoram. The solvents used in extracting and precipitating the spinosyn J and L mixture are recycled.
Spinosyns J and L, unlike spinosyns A and D, have a free hydroxyl group at the 30-position on the rhamnose sugar, which allows for chemical manipulation of this site (see Figure 5.10). In the first synthetic step, the free hydroxyl at the 30-position in spinosyn J and spinosyn L is ethylated to yield a mixture of 30-O-ethyl spinosyn J and 30-O-ethyl spinosyn L. This material is then hydrogenated to yield a mixture of spinetoram-J (30-O-ethyl-5,6-dihydro spinosyn J; see Figure 5.2, structure 5.5) and spinetoram-L (30-O-ethyl spinosyn L; see Figure 5.2, structure 5.6). The hydrogenation conditions are selective and reduce only the disubstituted double bond between C5 and C6 in the 30-O-ethyl spinosyn J intermediate, leaving the 30-O-ethyl spinosyn L unchanged. The material is crystallized from the reaction mixture and dried to create technical spinetoram, which is then formulated into end-use products.
5.9.3 Formulation Attributes of the Spinosyns
To meet a variety of market needs, spinosad and spinetoram products span a very wide range of formulation types (see Table 5.8).
The range of possible formulations for any pesticide is determined by the physical and chemical properties of the active ingredient. Three primary properties determine the formulation characteristics of the spinosyns: (1) bothFigure 5.10 Chemical synthesis steps in spinetoram manufacturing.
Table 5.8 Spinosyn product formulation types and associated uses.
Crops, ornamentals, forestry, stored grain, animal health, public health, turf, home and garden Public health Crops
Crops, ornamentals, seed treatment
Stored grain, crops
Crops
Crops, animal health, urban pests
Urban pests
Public health
Public health
Animal health
Urban pests
Public health are fermentation-derived mixtures; (2) both are weak bases; and (3) both have significant solubility in organic solvents.
As fermentation-derived products, spinosad and spinetoram are mixtures composed primarily of two similar, but not identical molecules. In terms of physical properties, a significant difference between the major and minor components of both spinosad and spinetoram is the presence or absence of a methyl group at C6 on the tetracycle (see Table 5.9). With regard to components of spinosad, spinosyn D (methyl group at C6) has a melting point 71 °C higher than that of spinosyn A (hydrogen at C6), and the water solubility of spinosyn D (at pH 7) is almost 1000-fold lower than that of spinosyn A. With regard to the components of spinetoram, spinetoram-L (methyl group at C6) has a melting point 72 °C lower than that of spinetoram-J (hydrogen at C6), and the water solubility of spinetoram-L (at pH 7) is four-fold higher than that of spinetoram-J. The melting points and water solubilities of the mixtures that constitute technical spinosad and technical spinetoram are determined by the relative ratios of the major and minor components.
The predominant components of both spinosad and spinetoram all have pKa values of about 8 (see Table 5.9). As a weak base, the solubility of spinosyns in water increases as the pH is reduced. From a formulation perspective, at pH level above 5, the spinosyns behave like high-melting solids with little water solubility, which results in the predominant agricultural formulations being suspension concentrates and wettable granule formulations composed of milled crystalline particles. Acid salts of spinosyns can be produced and are used in animal health formulations. The basic nature of the spinosyns is also a consideration when combining multiple active ingredients into the same formulation.
The spinosyns have significant solubility in organic solvents (see Table 5.9). This property is relatively rare in high-melting solids with limited water solubility, and has proven to be useful in a number of formulations for
Table 5.9 Selected physical properties of spinosyn A, spinosyn D, spinetoram-
J, and spinetoram-L.
Table 5.9 Selected physical properties of spinosyn A, spinosyn D, spinetoram-
J, and spinetoram-L.
Property
Spinosyn A133
Spinosyn D133
Spinetoram-J134
Spinetoram-L134
Melting point, °C
84-99.5a
161.6-170a
143.4b
70.8b
Water solubility,
235
0.332
11.3
46.7
mg/lc’d’e
pKaf
8.10e
7.87e
7.86g
7.59g
Solubility in organic solvents, mg/Lc
Acetone
168 000
10100
>250000
>250000
Ethyl acetate
194000
19 000
>250000
>250000
w-Heptane
12 400
300
23 900
>250000
Methanol
190000
2520
163 000
>250000
Xylene
> 250 000
64000
>250000
>250000
“Visual determination. bDiffential scanning calorimetry. cShake flask. ^Buffered to pH 7. eAt 20 °C.
fCapillary zone electrophoresis. gAt 25 °C.
“Visual determination. bDiffential scanning calorimetry. cShake flask. ^Buffered to pH 7. eAt 20 °C.
fCapillary zone electrophoresis. gAt 25 °C.
non-agricultural markets, such as mosquito control and animal health. It is also a consideration when combining the spinosyns with other active ingredients.
Two new related ways for the synthesis of lercanidipine have been reported: 1) The condensation of diketene (I) with the aminoalcohol (II) gives the corresponding acetoacetate ester (III), which is allowed to react with 3-nitrobenzaldehyde (IV) by means of HCl in chloroform yielding the expected benzylidene derivative (V). Finally, this compound is cyclized with methyl 3-aminocrotonate (VI) in refluxing isopropanol. 2) By esterification of 2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylic acid monomethyl ester (VIII) with alcohol (II) by means of SOCl2 in DMF/dichloromethane.
Aldeyra Therapeutics is developing reproxalap, which binds and traps free aldehydes, formulated using Captisol technology licensed from Ligand Pharmaceuticals as an eye drop formulation, for treating acute noninfectious anterior uveitis, allergic conjunctivitis and dry eye syndrome.
United States patent application serial number US 13/709,802, filed December 10, 2012 and published as US 2013/0190500 on July 25, 2013 (“the ‘500 publication,” the entirety of which is hereby incorporated herein by reference), describes certain aldehyde scavenging compounds. Such compounds include com ound A:
[0036] Compound A, (6-chloro-3-amino-2-(2-hydroxypropyl)-l-azanaphthalene), is designated as compound A in the ‘500 publication and the synthesis of compound A is described in detail at Example 5 of the ‘500 publication, and is reproduced herein for ease of reference.
Example A – General Preparation of Compound A
Compound A
[00436] The title compound was prepared according to the steps and intermediates (e.g., Scheme 1) described below and in the ‘500 publication, the entirety of which is incorporated herein by reference.
Step 1: Synthesis of Intermediate A- 1
[00437] To a 2 L round bottom flask was charged ethanol (220 mL), and pyridine (31 g, 392 mmol) and the resulting solution stirred at a moderate rate of agitation under nitrogen. To this solution was added ethyl bromopyruvate (76.6 g, 354 mmol) in a slow, steady stream. The reaction mixture was allowed to stir at 65±5° C. for 2 hours.
Step 2: Synthesis of Intermediate A-2
[00438] Upon completion of the 2-hour stir time in example 1, the reaction mixture was slowly cooled to 18-22° C. The flask was vacuum-purged three times at which time 2-amino-5-chloro-benzaldehyde (ACB) (50.0 g, 321 mmol) was added directly to the reaction flask as a solid using a long plastic funnel. Pyridine (64.0 g, 809 mmol) was added followed by an EtOH rinse (10 mL) and the reaction mixture was heated at 80±3° C. under nitrogen for about 16 hours (overnight) at which time HPLC analysis indicated that the reaction was effectively complete.
Step 3: Synthesis of Intermediate A-3
[00439] The reaction mixture from example 2 was cooled to about 70° C. and morpholine (76.0 g, 873 mmol)) was added to the 2 L reaction flask using an addition funnel. The reaction mixture was heated at 80±2° C. for about 2.5 hours at which time the reaction was considered complete by HPLC analysis (area % of A-3 stops increasing). The reaction mixture was cooled to 10-15° C. for the quench, work up, and isolation.
Step 4: Isolation of Intermediate A-3
[00440] To the 2 L reaction flask was charged water (600 g) using the addition funnel over 30-60 minutes, keeping the temperature below 15° C. by adjusting the rate of addition and using a cooling bath. The reaction mixture was stirred for an additional 45 minutes at 10-15° C. then the crude A-3 isolated by filtration using a Buchner funnel. The cake was washed with water (100 mLx4) each time allowing the water to percolate through the cake before applying a vacuum. The cake was air dried to provide crude A-3 as a nearly dry brown solid. The cake was returned to the 2 L reaction flask and heptane (350 mL) and EtOH (170 mL) were added and the mixture heated to 70±3° C. for 30-60 minutes. The slurry was cooled to 0-5° C. and isolated by filtration under vacuum. The A-3 was dried in a vacuum drying oven under vacuum and 35±3° C. overnight (16-18 hours) to provide A-3 as a dark green solid.
Step 5: Synthesis of Compound A
[00441] To a 2 L round bottom flask was charged methylmagnesium chloride (200 mL of 3.0 M solution in THF, 600 mmol). The solution was cooled to 0-5° C. using an ice bath.
[00442] A 500 mL flask (magnetic stirring) was charged with 22.8 grams A-3 from example 4 and THF (365 mL), stirred to dissolve then transferred to an addition funnel on the 2 L Reaction Flask. The A-3 solution was added drop-wise to the reaction flask over 5.75 hours, keeping the temperature of the reaction flask between 0-5° C throughout the addition. At the end of the addition the contents of the flask were stirred for an additional 15 minutes at 0-5° C. then the cooling bath was removed and the reaction was allowed to stir overnight at ambient temperature.
[00443] The flask was cooled in an ice bath and the reaction mixture was carefully quenched by adding EtOH (39.5 g, 857 mmol) drop-wise to the reaction mixture, keeping the temperature of the reaction mixture below 15° C. during the course of the addition. An aqueous solution of H4C1 (84.7 g H4C1 in 415 mL water) was then carefully added and the mixture stirred under moderate agitation for about 30 minutes then transferred to a separately funnel to allow the layers to separate. Solids were present in the aqueous phase so HO Ac (12.5 g) was added and the contents swirled gently to obtain a nearly homogeneous lower aqueous phase. The lower aqueous layer was transferred back to the 2 L reaction flask and stirred under moderate agitation with 2-methylTHF (50 mL) for about 15 minutes. The original upper organic layer was reduced in volume to approximately 40 mL using a rotary evaporator at≤40° C. and vacuum as needed. The phases in the separatory funnel were separated and the upper 2-MeTHF phase combined with the product residue, transferred to a 500 mL flask and vacuum distilled to an approximate volume of 25 mL. To this residue was added 2-MeTHF (50 mL) and distilled to an approximate volume of 50 mL. The crude compound A solution was diluted with 2-MeTHF (125 mL), cooled to 5-10° C. and 2M H2S04 (aq) (250 mL) was slowly added and the mixture stirred for 30 minutes as the temperature was allowed to return to ambient. Heptane (40 mL) was charged and the reaction mixture stirred for an additional 15 minutes then transferred to a separatory funnel and the layers were allowed to separate. The lower aqueous product layer was extracted with additional heptane (35 mL) then the lower aqueous phase was transferred to a 1 L reaction flask equipped with a mechanical stirrer and the mixture was cooled to 5-10° C. The combined organic layers were discarded. A solution of 25% NaOH(aq) was prepared (NaOH, 47 g, water, 200 mL) and slowly added to the 1 L reaction flask to bring the pH to a range of 6.5-8.5.
[00444] EtOAc (250 mL) was added and the mixture was stirred overnight. The mixture was transferred to a separatory funnel and the lower phase discarded. The upper organic layer was washed with brine (25 mL) then the upper organic product layer was reduced in volume on a rotary evaporator to obtain the crude compound A as a dark oil that solidified within a few minutes. The crude compound A was dissolved in EtOAc (20 mL) and filtered through a plug of silica gel (23 g) eluting with 3/1 heptane/EtOAc until all compound A was eluted (approximately 420 mL required) to remove most of the dark color of compound A. The solvent was removed in vacuo to provide 14.7 g of compound A as a tan solid. Compound A was taken up in EtOAc (25 mL) and eluted through a column of silica gel (72 g) using a mobile phase gradient of 7/1 heptane/EtOAc to 3/lheptane/EtOAc (1400 mL total). The solvent fractions containing compound A were stripped, compound A diluted with EtOAc (120 mL) and stirred in a flask with Darco G-60 decolorizing carbon (4.0 g) for about 1 hour. The mixture was filtered through celite using a fitted funnel, rinsing the cake with EtOAc (3 x 15 mL). The combined filtrates were stripped on a rotary evaporator and compound A dissolved in heptane (160 mL)/EtOAc(16 mL) at 76° C. The
homogeneous solution was slowly cooled to 0-5° C, held for 2 hours then compound A was isolated by filtration. After drying in a vacuum oven for 5 hours at 35° C. under best vacuum, compound A was obtained as a white solid. HPLC purity: 100% (AUC).
Example 1 – Preparation of Free Base Forms A and B of Compound A
Compound A
[00445] Compound A is prepared according to the method described in detail in Examples 1-5 of the ‘500 publication, the entirety of which is hereby incorporated herein by reference.
[00190] 2-(3-amino-6-chloroquinolin-2-yl)propan-2-ol. To a 2 L round bottom flask was charged methylmagnesium chloride (200 mL of 3.0 M solution in THF, 600 mmol). The solution was cooled to 0-5 °C using an ice bath.
[00191] A 500 mL flask (magnetic stirring) was charged with 22.8 grams A-3a from Example 4 and THF (365 mL), stirred to dissolve, and then transferred to an addition funnel on the 2 L reaction flask. The A-3a solution was added drop-wise to the reaction flask over 5.75 hours, keeping the temperature of the reaction flask between 0-5 °C throughout the addition. At the end of the addition the contents of the flask were stirred for an additional 15 minutes at 0-5 °C, then the cooling bath was removed and the reaction was allowed to stir overnight at ambient temperature.
[00192] The flask was cooled in an ice bath and the reaction mixture was carefully quenched by adding EtOH (39.5 g, 857 mmol) drop-wise to the reaction mixture, keeping the temperature of the reaction mixture below 15 °C during the course of the addition. An aqueous solution of H4CI (84.7 g H4CI in 415 mL water) was then carefully added and the mixture stirred under moderate agitation for about 30 minutes then transferred to a separatory funnel to allow the layers to separate. Solids were present in the aqueous phase so HOAc (12.5 g) was added and the contents swirled gently to obtain a nearly homogeneous lower aqueous phase. The lower aqueous layer was transferred back to the 2 L reaction flask and stirred under moderate agitation with 2-methyl-tetrahydrofuran (2-MeTHF) (50 mL) for about 15 minutes. The original upper organic layer was reduced in volume to approximately 40 mL using a rotary evaporator at < 40 °C under vacuum as needed. The phases in the separatory funnel were separated and the upper 2-MeTHF phase combined with the product residue was transferred to a 500 mL flask and vacuum distilled to an approximate volume of 25 mL. To this residue was added 2-MeTHF (50 mL) and the mixture again distilled to an approximate volume of 50 mL. The crude compound NS2 solution was diluted with 2-MeTHF (125 mL), cooled to 5-10 °C, and 2 M H2S04 (aq) (250 mL) was slowly added and the mixture stirred for 30 minutes as the temperature was allowed to return to ambient. Heptane (40 mL) was charged and the reaction mixture stirred for an additional 15 minutes then transferred to a separatory funnel, and the layers were allowed to separate. The lower aqueous product layer was extracted with additional heptane (35 mL), then the lower aqueous phase was transferred to a 1 L reaction flask equipped with a mechanical stirrer, and the mixture was cooled to 5-10 °C. The combined organic layers were discarded. A solution of 25% NaOH (aq) was prepared (NaOH, 47 g, water, 200 mL) and slowly added to the 1 L reaction flask to bring the pH to a range of 6.5 – 8.5.
[00193] EtOAc (250 mL) was added and the mixture was stirred overnight. The mixture was transferred to a separatory funnel and the lower phase discarded. The upper organic layer was washed with brine (25 mL), then the upper organic product layer was reduced in volume on a rotary evaporator to obtain a obtain the crude compound NS2 as a dark oil that solidified within a few minutes. The crude compound NS2 was dissolved in EtOAc (20 mL) and filtered through a plug of silica gel (23 g) eluting with 3/1 heptane/EtOAc until all compound NS2 was eluted (approximately 420 mL required) to remove most of the dark color of compound NS2. The solvent was removed in vacuo to provide 14.7 g of compound NS2 as a tan solid. Compound NS2 was taken up in EtOAc (25 mL) and eluted through a column of silica gel (72g) using a mobile phase gradient of 7/1 heptane/EtOAc to 3/1 heptane/EtOAc (1400 mL total). The solvent fractions containing compound NS2 were evaporated. Compound NS2 was diluted with EtOAc (120 mL) and stirred in a flask with Darco G-60 decolorizing carbon (4.0 g) for about 1 hour. The mixture was filtered through celite using a firtted funnel, rinsing the cake with EtOAc (3 x 15 mL). The combined filtrates were evaporated on a rotary evaporator and compound NS2 dissolved in heptane (160 mL)/EtOAc (16 mL) at 76 °C. The homogeneous solution was slowly cooled to 0-5 °C, held for 2 hours, then compound NS2 was isolated by filtration. After drying in a vacuum oven for 5 hours at 35 °C under best vacuum, compound NS2 was obtained as a white solid. HPLC purity: 100% (AUC); HPLC (using standard conditions): A-2: 7.2 minutes; A-3 : 11.6 minutes.
Preparation of ACB
[00194] After a N2 atmosphere had been established and a slight stream of N2 was flowing through the vessel, platinum, sulfided, 5 wt. % on carbon, reduced, dry (9.04 g, 3.0 wt. % vs the nitro substrate) was added to a 5 L heavy walled pressure vessel equipped with a large magnetic stir-bar and a thermocouple. MeOH (1.50 L), 5-chloro-2-nitrobenzaldehyde (302.1 g, 1.63 mol), further MeOH (1.50 L) and Na2C03 (2.42 g, 22.8 mmol, 0.014 equiv) were added. The flask was sealed and stirring was initiated at 450 rpm. The solution was evacuated and repressurized with N2 (35 psi), 2x. The flask was evacuated and repressurized with H2 to 35 psi. The temperature of the solution reached 30 °C w/in 20 min. The solution was then cooled with a water bath. Ice was added to the water bath to maintain a temperature below 35 °C. Every 2h, the reaction was monitored by evacuating and repressurizing with N2 (5 psi), 2x prior to opening. The progress of the reaction could be followed by TLC: 5-Chloro-2-nitrobenzaldehyde (Rf = 0.60, CH2CI2, UV) and the intermediates (Rf = 0.51, CH2CI2, UV and Rf = 0.14, CH2CI2, UV) were consumed to give ACB (Rf = 0.43, CH2CI2, UV). At 5 h, the reaction had gone to 98% completion (GC), and was considered complete. To a 3 L medium fritted funnel was added celite (ca. 80 g). This was settled with MeOH (ca. 200 mL) and pulled dry with vacuum. The reduced solution was transferred via cannula into the funnel while gentle vacuum was used to pull the solution through the celite plug. This was chased with MeOH (4 x 150 mL). The solution was transferred to a 5 L three-necked round-bottom flask. At 30 °C on a rotavap, solvent (ca. 2 L) was removed under reduced pressure. An N2 blanket was applied. The solution was transferred to a 5L four-necked round-bottomed flask equipped with mechanical stirring and an addition funnel. Water (2.5 L) was added dropwise into the vigorously stirring solution over 4 h. The slurry was filtered with a minimal amount of vacuum. The collected solid was washed with water (2 x 1.5 L), 2-propanol (160 mL) then hexanes (2 x 450 mL). The collected solid (a canary yellow, granular solid) was transferred to a 150 x 75 recrystallizing dish. The solid was then dried under reduced pressure (26-28 in Hg) at 40°C overnight in a vacuum-oven. ACB (> 99% by HPLC) was stored under a N2 atmosphere at 5°C.
PATENT
WO-2020223717
Process for preparing reproxalap as acetaldehyde dehydrogenase inhibitor useful for treating ocular diseases and cancer.
PATENT
WO-2020223685
Novel crystalline forms of reproxalap (compound 1; designated as Forms A and B) as acetaldehyde dehydrogenase inhibitor useful for treating ocular diseases and cancer.
Keoxifene hydrochloride, Raloxifene hydrochloride, LY-139481(free base), LY-156758, Optruma, Loxifen, EvistaTitle: RaloxifeneCAS Registry Number: 84449-90-1CAS Name: [6-Hydroxy-2-(4-hydroxyphenyl)benzo[b]thien-3-yl][4-[2-(1-piperidinyl)ethoxy]phenyl]methanoneAdditional Names: keoxifeneManufacturers’ Codes: LY-139481Molecular Formula: C28H27NO4SMolecular Weight: 473.58Percent Composition: C 71.01%, H 5.75%, N 2.96%, O 13.51%, S 6.77%Literature References: Nonsteroidal, selective estrogen receptor modulator (SERM). Prepn: C. D. Jones, EP62503; idem,US4418068 (1982, 1983 both to Lilly); idemet al.,J. Med. Chem.27, 1057 (1984). Review of pharmacology and toxicology: J. Buelke-Sam et al.,Reprod. Toxicol.12, 217-221 (1998); of clinical pharmacology and pharmacokinetics: D. Hochner-Celnikier, Eur. J. Obstet. Gynecol. Reprod. Biol.85, 23-29 (1999); of clinical efficacy in osteoporosis: D. Agnusdei, ibid. 43-46. Clinical effect on risk of breast cancer: S. R. Cummings et al.,J. Am. Med. Assoc.281, 2189 (1999); on reduction of fracture risk: B. Ettinger et al.,ibid.282, 637 (1999).Properties: Crystals from acetone, mp 143-147°. uv max (ethanol): 290 nm (e 34000).Melting point: mp 143-147°Absorption maximum: uv max (ethanol): 290 nm (e 34000) Derivative Type: HydrochlorideCAS Registry Number: 82640-04-8Manufacturers’ Codes: LY-156758Trademarks: Evista (Lilly)Molecular Formula: C28H27NO4S.HClMolecular Weight: 510.04Percent Composition: C 65.94%, H 5.53%, N 2.75%, O 12.55%, S 6.29%, Cl 6.95%Properties: Crystals from methanol/water, mp 258°. uv max (ethanol): 286 nm (e 32800).Melting point: mp 258°Absorption maximum: uv max (ethanol): 286 nm (e 32800) Therap-Cat: Antiosteoporotic.Keywords: Antiosteoporotic; Selective Estrogen Receptor Modulator (SERM).
Raloxifene was approved for medical use in the United States in 1997.[4] It is available as a generic medication.[4][6] A month supply in the United Kingdom costs the NHS about 3.50 £ as of 2019.[6] In the United States the wholesale cost of this amount is about $16.[7] In 2017, it was the 330th most commonly prescribed medication in the United States, with more than 900 thousand prescriptions.[8
Medical uses
Raloxifene is used for the treatment and prevention of osteoporosis in postmenopausal women.[9] It is used at a dosage of 60 mg/day for both the prevention and treatment of osteoporosis.[10] In the case of either osteoporosis prevention or treatment, supplemental calcium and vitamin D should be added to the diet if daily intake is inadequate.[11]
Raloxifene is used to reduce the risk of breast cancer in postmenopausal women. It is used at a dosage of 60 mg/day for this indication.[10] In the Multiple Outcomes of Raloxifene (MORE) clinical trial, raloxifene decreased the risk of all types of breast cancer by 62%, of invasive breast cancer by 72%, and of invasive estrogen receptor-positive breast cancer by 84%.[12] Conversely, it does not reduce the risk of estrogen receptor-negative breast cancer.[12] There were no obvious differences in effectiveness of raloxifene in the MORE trial for prevention of breast cancer at a dosage of 60 mg/m2/day relative to 120 mg/m2/day.[12] In the Study of Tamoxifen and Raloxifene (STAR) trial, 60 mg/day raloxifene was 78% as effective as 20 mg/day tamoxifen in preventing non-invasive breast cancer.[13] Women with undetectable levels of estradiol (<2.7 pg/mL) have a naturally low risk of breast cancer and, in contrast to women with detectable levels of estradiol, do not experience significant benefit from raloxifene in terms of reduction of breast cancer risk.[12]
Raloxifene may infrequently cause serious blood clots to form in the legs, lungs, or eyes.[1] Other reactions experienced include leg swelling/pain, trouble breathing, chest pain, and vision changes. Black box warnings were added to the label of raloxifene in 2007 warning of increased risk of death due to stroke for postmenopausal women with documented coronary heart disease or at increased risk for major coronary events, as well as increased risk for deep vein thrombosis and pulmonary embolism.[14] The risk of venous thromboembolism with raloxifene is increased by several-fold in postmenopausal women (RR = 3.1).[18][12] Raloxifene has a lower risk of thromboembolism than tamoxifen.[13] In the MORE trial, raloxifene caused a 40% decrease in risk of cardiovascular events in women who were at increased risk for coronary artery disease, although there was no decrease in cardiovascular events for the group as a whole.[12]
A report in September 2009 from Health and Human Services’ Agency for Healthcare Research and Quality suggests that tamoxifen and raloxifene, used to treat breast cancer, significantly reduce invasive breast cancer in midlife and older women, but also increase the risk of adverse side effects.[19]
A recent human case report in July 2016 suggests that raloxifene may in fact, at some point, also stimulate breast cancer growth leading to a reduction of advanced breast cancer disease upon the withdrawal of the drug.[20]
Raloxifene is metabolized in the liver and undergoes enterohepatic recycling.[2] It is metabolized exclusively by glucuronidation and is not metabolized by the cytochrome P450 system.[1][2] Less than 1% of radiolabeled material in plasma comprises unconjugated raloxifene.[2] The metabolites of raloxifene include several glucuronides.[1] The elimination half-life of raloxifene after a single dose is 27.7 hours (1.2 days), whereas its half-life at steady state at a dosage of 60 mg/day is 15.8 to 86.6 hours (0.7–3.6 days), with an average of 32.5 hours (1.4 days).[1][2] The extended half-life of raloxifene is attributed to enterohepatic recirculation and its high plasma protein binding.[1] Raloxifene and its glucuronideconjugates are interconverted by reversible metabolism and enterohepatic recycling, which prolongs the elimination half-life of raloxifene with oral administration.[2] The medication is deconjugated into its active form in a variety of tissues, including liver, lungs, spleen, bone, uterus, and kidneys.[1]
Elimination
Raloxifene is mainly excreted in bile and is eliminated in feces.[1][2] Less than 0.2% of a dose is excreted unchanged in urine and less than 6% of a dose is excreted in urine as glucuronide conjugates.[2]
Raloxifene hydrochloride has the empirical formula C28H27NO4S•HCl, which corresponds to a molecular weight of 510.05 g/mol. Raloxifene hydrochloride is an off-white to pale-yellow solid that is slightly soluble in water.[14]
Raloxifene was approved in the United States for the prevention of postmenopausal osteoporosis in 1997, the treatment of postmenopausal osteoporosis in 1999, and to prevent or reduce the risk of breast cancer in certain postmenopausal women in 2007.[39][40][41][42] It received orphan designation in 2005.[39]
Raloxifene is sold mainly under the brand name Evista and to a lesser extent the brand name Optruma.[46][44] It is also sold under a variety of other brand names in various countries.[46]
Raloxifene is provided in the form of 60 mg oraltablets.[10]
Controversy
An editorial in Lancet Oncology criticized the way that research about the medication for breast cancer prevention was released.[47]
Research
Clinical studies of raloxifene for metastatic breast cancer in women have been conducted but found little effectiveness at 60 mg/day in those previously treated with tamoxifen, though modest effectiveness has been observed at higher doses.[12][48] In contrast to tamoxifen, raloxifene is not approved for the treatment of breast cancer.[49]
Raloxifene has been studied as an adjunct in the treatment of schizophrenia in postmenopausal women.[59] A 2017 meta-analysis concluded that it was safe and effective for this indication, although further studies with larger sample sizes are needed for confirmation.[59] It may be effective in women with less severe symptoms.[59]
A tissue-selective estrogen-receptor complex (TSEC) of estradiol and raloxifene has been studied in postmenopausal women.[60]
June 18th 2020, Exscalate4CoV, the private-public consortium supported by the EU’s Horizon 2020 programme for research and innovation, led by Dompé farmaceutici and currently representing 18 partners (including Fraunhofer Institute, CINECA, Chelonia Applied Science, Swiss Institute of Bioinformatics and others) has requested access to clinical trials for the use of Raloxifene in Covid 19 patients. Raloxifene, already proven effective against Mers and Sars in precliinical tests, has been indicated as effective against Sars-Cov2 by the “in-silico” research conducted by the consortium which has shown efficacy in countering the replication of the virus in cells. The IP for its use against Sars-Cov2 has already been protected on May 6 2020 in the name Dompé farmaceutici, Fraunhofer Institute and KU Leuven, to facilitate the largest possible access. Raloxifene would be used in mildly symptomatic Covid19 patients to halt the spread of infection. This result emerged from the first virtual (in silico) screening conducted on the Consortium’s supercomputers of more than 400.000 molecules (safe-in-man drugs and natural products) made available by Dompé farmaceutici and the partner Fraunhofer (IME) to the Consortium. The molecules were prioritized if in clinical stage or already on the market. 7.000 molecules with certain promising characteristics were tested.
SYN
Jones, Charles D.; Jevnikar, Mary G.; Pike, Andrew J.; Peters, Mary K.; Black, Larry J.; Thompson, Allen R.; Falcone, Julie F.; Clemens, James A. (1984). “Antiestrogens. 2. Structure-activity studies in a series of 3-aroyl-2-arylbenzo[b]thiophene derivatives leading to [6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thien-3-yl]-[4-[2-(1-piperidinyl)ethoxy]phenyl]methanone hydrochloride (LY 156758), a remarkably effective estrogen antagonist with only minimal intrinsic estrogenicity”. Journal of Medicinal Chemistry27 (8): 1057–66.doi:10.1021/jm00374a021. PMID6431104.
syn 1
EP 0062053; GB 2097788
Keoxifene has been synthesized using the following process: A portion of 6-methanesulfonyloxy-2-(4-methanesulfonyloxyphenyl)-3-[4-(2-pipendinoethoxy)benzoyl]benzo[b]thiophene hydrochloride (I) was combined with denatured alcohol and 5N sodium hydroxide, and stirred under a nitrogen atmosphere. The reaction mixture was evaporated to dryness under vacuum, and the residue dissolved in water and washed with diethyl ether. The water layer was degassed under vacuum, and then nitrogen was bubbled through it to remove all traces of ether. The mixture was then acidified with 1N hydrochloric acid, and then made basic with excess sodium bicarbonate The precipitate was collected by filtration and washed with cold water to obtain crude product, which was purified on a column of silica gel. The column was eluted first with 700 ml of 5% methanol in chloroform, followed by 1l of 10% methanol in chloroform. The impurities came off first, and the product-containing fractions were combined and evaporated under vacuum to obtain a yellow oil. The oil was dissolved in acetone seeded and chilled in a freezer to obtain the purified product.
syn2
J Label Compd Radiopharm 1995,36(1),43
The synthesis of radiolabeled raloxifene has been reported: The esterification of 3,5-dibromo-4-hydroxybenzoic acid (I) with methanol/HCl gives the corresponding methyl ester (II), which is condensed with 1-(2-chloroethyl)piperidine (III) by means of K2CO3 in DMF yielding 3,5-dibromo-4-[2-(1-piperidyl)ethoxy]benzoic acid methyl ester (IV). The hydrolysis of (IV) with NaOH in methanol affords the corresponding free acid (V), which by treatment of SOCl2 in toluene is converted to the acyl chloride (VI). The Friedel-Crafts condensation of (VI) with 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene (VII) by means of AlCl3 in dichloromethane gives [3,5-dibromo-4-[2-(1-piperidinyl)ethoxy]phenyl]-[6-methoxy-2-(4-methoxy phenyl)benzo[b]thien-3-yl]methanone (VIII), which is demethylated with AlCl3 and ethylmercaptane to dibromoraloxifene (IX). Finally, this compound is submitted to hydrogenolysis with tritium over Pd/C in methanol.
syn 3
Bioorg Med Chem Lett 1997,7(8),993
The two major metabolites of raloxifene, the glucuronide conjugates (VI) and (VIII) are synthesized as follows: The partial silylation of raloxifene (I) with tert-butyldimethylsilyl chloride (TBDMS-Cl) by means of dimethylaminopyridine (DMAP) in THF/DMF gives a mixture of the monosilylated compounds (II) and (III), which are separated by chromatography. Compounds (II) and (III) are independently condensed with methyl 1,2,3,4-tetra-O-acetyl-D-glucuronate (IV) by means of BF3.OEt2 in dichloromethane yielding protected glucuronides (V) and (VII), respectively. Finally, both compounds are deprotected by a treatment first with LiOH in dioxane to hydrolyzed the ester groups, and then with tetrabutylammonium fluoride in THF to eliminate the silyl groups, thus obtaining the desired metabolites (VI) and (VIII), respectively.
syn 4
Tetrahedron Lett 1999,40(28),5155
Two related new syntheses of raloxifene have been described: 1) The acylation of N-(6-methoxy-1-benzothiophen-2-yl)-N,N-dimethylamine (I) with 4-fluorobenzoyl chloride (II) by heating at 100 C in chlorobenzene gives the 3-acyl derivative (III), which is condensed with 4-methoxyphenylmagnesium bromide (IV) in THF yielding 3-(4-fluorobenzoyl)-6-methoxy-2-(4-methoxyphenyl)-1-benzothiophene (V). The condensation of (V) with 1-(2-hydroxyethyl)piperidine (VI) by means of NaH in DMF affords the ether (VII), which is finally demethylated with AlCl3 and ethanethiol. 2) The intermediate (III) can also be condensed first with 1-(2-hydroxyethyl)piperidine (VI) by means of NaH as before giving the piperidinoethyl ether (VIII), which is then condensed with the Grignard reagent (IV) affording the previously reported ether (VII).
A GREEN PROCESS FOR DEMETHYLATION REACTION IN SYNTHESIS OF RALOXIFENE HYDROCHLORIDEAuthors : Ramadas Chavakula *, Chakradhar Saladi J S, Narayana Rao Mutyalaa , Vijaya Raju Maddalaa and Raghu Babu Kb
A green process for demethylation reaction in synthesis of raloxifene hydrochloride by using aluminium chloride and odorless decanethiol as demethylation agent instead of aluminium chloride and ethanethiol (foul smell) under normal conditions is described.
Raloxifene hydrochloride [1], is an estrogen agonist/antagonist, commonly referred to as a Selective Estrogen Receptor Modulator (SERM) [1,2] that belongs to the benzothiophene class of compounds. Raloxifene decreases the resorption of bone and reduces the biochemical markers of bone turnover to the premenopausal range [3–5]. Raloxifene hydrochloride may also lower the chance of developing a certain type of breast cancer (invasive breast cancer) in post-menopausal women [6,7]. It can be synthesized [3] directly from aroylation of 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene [2] by the acid chloride(4) of 4-[2-(1-piperidinyl)ethoxy]benzoic acid hydrochloride [3] in the presence of AlCl3 followed by addition of ethanethiol (FIG. 1).
Experimental Section
4-[2-(1-Piperidinyl)ethoxy]benzoic acid hydrochloride [3] and 6-methoxy-2-(4-methoxyphenyl) benzo[b] thiophene [2] were prepared by procedures reported previously [3]. Decanethiol was from commercial source. All melting points are uncorrected and were determined in capillary tubes on an Electothermal melting point apparatus. 1H NMR spectra were recorded on a Brucker ADVANCE 400 MHz spectrometer, using DMSO-d6 as solvent and TMS as internal standard. Electrospray ionization mass spectroscopy was performed using an ion trap mass spectrometer (Model 6310 Agilent). All reactions were monitored and checked by Thin Layer Chromatography (TLC) using methanol and spots examined by a UV lamp.
Preparation of [6-hydroxy-2-(4-hydroxyphenyl)benzo[b]thiophen-3-yl][4-[2-(1-piperidyl)ethoxy]phenyl] methanone hydrochloride (Raloxifene hydrochloride) [1]
To a solution of 4-[2-(1-piperidinyl)ethoxy]benzoic acid hydrochloride (3) (14.3 g, 0.05 mol) in methylene dichloride (400 mL) and pyridine (0.5 mL) at 25ºC to 35ºC, thionyl chloride (23.8 g, 0.20 mol) was added dropwise under argon for 15-30 minute. The reaction mixture was stirred for 2 hr. at 40ºC to 45ºC. Excess thionyl chloride and solvent were removed in vacuum at 40◦C to afford 15.0 g of the crude acid chloride hydrochloride salt [4]. The crude solid acid chloride hydrochloride [4] was dissolved in methylene dichloride (150 mL), cooled to 0ºC to 10ºC, 6-methoxy-2-(4-methoxyphenyl)benzo[b] thiophene [2] (10.8 g, 0.04 mol) was added. Then, anhydrous aluminium chloride (37.0 g, 0.28 mol) was added portion wise over a period of 30 min and then the mixture was allowed to warm to 30ºC and stirred for 2 hr at 25-35ºC. Then decanethiol (28.0 g, 0.16 mol) was added and stirred for 2 hr. at 25-35ºC. The reaction mixture was quenched with mixture of methanol (100 mL), ice (200 g) and Conc. HCl (15 mL) and stirred for 1 hr. at 25-35ºC. The precipitated solid was collected, washed with water (100 mL X 2) and dried at 65ºC for 4 h to afford 20.0 g of crude compound 1, which was crystallized from methanol/water (23/1, vol/vol) to yield 13.6 g of compound 1 (53.3 %yield) as a white solid, MP 258-260°C, liter 3, 258°C ; 1H NMR: δ 1.34, 1.72 [2H, m, (CH2CH2)2CH2], 1.76 [4H, m, N(CH2CH2)2], 2.96 (2H, m, N-CH2), 3.43 [4H, m, N(CH2CH2)2], 4.44 (2H, m, O-CH2), 6.67 (2H, d, Ar), 6.85 (1H, d, Ar), 6.95 (2H, d, Ar), 7.18 (2H, d, Ar), 7.25 (1H, d, Ar), 7.35 (1H, s, Ar), 7.70 (2H, d, Ar), 9.77 (1H, s, OH), 9.82 (1H, s, OH), 10.16 (1H, brs, NH), MS (ESI): m/z 474.6 (M +H). “This procedure has been scaled up using 250g of compound 1.”
Results and Discussion
Commonly used thiols like ethanethiol and benzyl mercaptan in demethylation reactions have a foul smell making them difficult and unpleasant to use in the laboratory without fume hoods. The problem becomes even worse in industry on a large scale. Odorless substitutes are therefore always required. Few papers [8,9] discuss the use of long chain thiols to minimize odor, so we used this work as a basis for choosing a long chain thiol for our demethylation reaction. We now report a new, highly active demethylation reagent, an aluminum chloride and decanethiol, characterized by rapid action under mild conditions, easy workup of the reaction product, and high yield (FIG. 2.).
Figure 2: Synthesis of Raloxifene hydrochloride.
Conclusion
In conclusion, we have found that decanethiol is odorless thiol compared to ethanethiol. We believe that removing the foul-smelling thiols and use of these odorless thiols will greatly improve the greenchemistry.
Bryant HU, Dere WH. Selective estrogen receptor modulators: an alternative to hormone replacement therapy. Proc Soc Exp Biol Med. 1998;217:45-52.
Jones CD, Jevnikar MG, Pike AJ, et al. Antiestrogens. 2. Structure-activity studies in a series of 3-aroyl-2-arylbenzo [b] thiophene derivatives leading to [6-hydroxy-2-(4-hydroxyphenyl) benzo [b] thien-3-yl]-[4-[2-(1-piperidinyl) ethoxy] phenyl] methanone hydrochloride (LY 156758), a remarkably effective estrogen antagonist with only minimal intrinsic estrogenicity. J Med Chem. 1984;27:1057-66.
Sato M, Grese TA, Dodge JA, et al. Emerging therapies for the prevention or treatment of postmenopausal osteoporosis. J Med Chem. 1999;42:1-24.
Draper MW, Flowers DE, Huster WJ, et al. A controlled trial of raloxifene (LY139481) HCl: impact on bone turnover and serum lipid profile in healthy postmenopausal women. J Bone Miner Res. 1996;11:835-42.
Mild and high-yielding synthesis is described for raloxifene via piperdine nucleophilic substitution of a new raloxifene intermediate 3-aroyl-2-aryl-substituted benzo[b]thiophenes, which is obtained by acylation of para-substituted benzoyl chlorides and 2-arylbenzo[b]thiophenes. The key step is solvent free and offers valuable advantages, such as low cost, and is suitable for industrial production.
The improved synthesis of raloxifene 1 was accomplished as shown in Scheme 2. Methyl p-hydroxybenzoate 2, 1-bromo-2-chloroethane, and K2CO3 were refluxed in acetone, yielding compound 3 in 94% yield. Without prior purification, 3 was hydrolyzed to the corresponding p-substituted benzoyl acids 4 in 100% yield. The application of general reaction conditions of methanol as solvent and hydrochloric as acid would afford the substitution impurity 4-(2-methoxyethoxy)-benzoic acid. To control this impurity during reaction, various solvents such as alcohol, ethyl acetate, acetone, and tetrahydrofuran (THF) were screened, and THF gave the best result from the view of impurity formation and yield. Compound 4 is a solid and was easily isolated from THF by adding water. Then 4 was transferred to acid chlorides 5 and substantially reacted with benzothiophene 6 using AlCl3 in dichloromethane at 50 C to afford aroylated benzothiophene 7 in two steps, with yield of 95% (79% from method A[8] and 65.5% from method B[3]). With the requisite 7 in hand, we next examined piperidine nucleophilic substitution to produce the desired beno[b]thien-3-yl ketones 8. In general using reaction conditions A (acetone, NaI, K2CO3, reflux, 70%) and B (acetonitrile, NaI, K2CO3, reflux, 85%), impurity formation was observed from the beginning of the reaction. We screened various conditions and were delighted to found that using excess piperidine at reflux temperature gave negligible impurity formation. Piperidine was not only reagent but also solvent. The isolated product 8 was stable and was converted into the desired raloxifene 1 as reported. In conclusion, we have developed a viable alternative route for the synthesis of raloxifene. The new synthesis would have been better able to support the increase in bulk demand for this drug for the chemoprevention of breast cancer and novel formulations. Our synthetic route has several advantages: the use of difunctionalized coumpunds 5 as key intermediate makes Friedel–Crafts acylation and nucleophilic substitution highly efficient. The using of piperine as reagent and solvent avoids the large waste streams derived from neutralization reaction of sodium hydride. The cost of the new route is less than the current route of manufacture.
Preparation of [4-(2-Chloro-ethoxy)-phenyl]-[6-methoxy-2- (4-methoxy-phenyl)-benzo[b]thiophen-3-yl]-methanone (7) Under an N2 atmosphere, 5 was added to a mixture of 6 (20.25 g, 75 mmol) and AlCl3 (13.30 g, 100 mmol) in DCM (2 mL), and the mixture was stirred for 12 h. The reaction was monitored by TLC (n-hexane/EtOAc, 4:1). After the reaction was judged complete, the reaction mixture was allowed to cool. The crude mixture was poured into H2O and extracted with EtOAc. The organic layer was separated and concentrated. The residue was crystallized from EtOAc to give the product 7 (32.26 g, 95%): yellow solid crystals; mp 119–120 C; IR (KBr) nmax: 2960, 2835, 1647, 1599, 1472, 1251, 1169, 1032, 830 cm1 ; 1 H NMR (400 MHz, CDCl3) d 7.76 (d, J ¼ 8.8 Hz, 2H), 7.53 (d, J ¼ 8.8 Hz, 1H), 7.32 (d, J ¼ 8.4 Hz, 2H), 7.31 (s, 1H), 6.95 (dd, J ¼ 8.4, 2.4 Hz, 1H), 6.75 (dd, J ¼ 9.2, 7.2 Hz, 4H), 4.20 (t, J ¼ 4.0 Hz,2H), 3.87 (s, 3H), 3.78 (t, J ¼ 6.0 Hz, 2H), 3.74 (s, 3H); 13C NMR (100 MHz, CDCl3) d 193.1, 162.1, 159.7, 157.6, 142.7, 139.9, 133.8, 132.3, 130.9, 130.2, 130.1, 125.9, 123.9, 114.8, 114.1, 113.9, 104.4, 67.8, 55.6, 55.2, 41.5; MS (EI) m/z (%):452 (Mþ, 100.0), 437 (13.0), 297 (25.0), 183 (39.0), 121 (44.0). HRMS m/z (EI) calcd. for C25H22ClO4S: (MþH) þ: 453.0927; found: 453.0933. Preparation of [6-Methoxy-2-(4-methoxy-phenyl)-benzo[b] thiophen-3-yl]-[4-(2-piperidin-1-yl-ethoxy)-phenyl]-methanone (8) Under an N2 atmosphere, a mixture of 7 (8.50 g, 19 mmol) and piperdine (30 ml) was stirred under reflux for 12 h. The reaction was monitored by TLC (n-hexane/EtOAc, 4:1). After the reaction was judged complete, the reaction mixture was allowed to cool. The mixture was concentrated for recovery of piperidine. EtOAc was added and the residue was washed with saturated NaHCO3 aqueous solution. The organic layer was separated and concentrated to give the product 8 (8.80 g, 94%): yellow viscous oil; IR (KBr) nmax: cm1 2933, 1645, 1597, 1535, 1501, 1470, 1249, 1164, 1030, 827; 1 H NMR (400 MHz, CDCl3) d7.76 (d, J ¼ 8.8 Hz, 2H), 7.52 (d, J ¼ 8.8 Hz, 1H), 7.33 (d, J ¼ 8.8 Hz, 2H), 7.30 (d, J ¼ 2.4 Hz, 1H), 6.94 (dd, J ¼ 8.8, 2.0 Hz, 1H), 6.75 (dd, J ¼ 7.2, 5.2 Hz, 4H), 4.08 (t, J ¼ 6.0 Hz, 2H), 3.86 (s, 3H), 3.73 (s, 3H), 2.71 (t, J ¼ 6.0 Hz, 2H), 2.46 (s, 4H), 1.60–1.54 (m, 4H), 1.43–1.41 (m, 2H).13C NMR (100 MHz, CDCl3) d 193.2, 163.0, 159.7, 157.6, 142.4, 140.1, 133.9, 132.3, 130.6, 130.4, 130.2, 126.0, 124.0, 114.8, 114.2, 114.1, 104.5, 66.3, 57.7, 55.6, 55.2, 55.1, 25.9, 24.1. MS (EI) m/z (%): 501 (Mþ, 100.0), 452 (12.0), 402 (21.0), 297 (24.0), 98 (100.0). HRMS m/z (EI) calcd. for C30H32NO4S: (MþH) þ: 502.2052; found: 502.2055.REFERENCES 1. Clemett, D.; Spencer, C. M. Drugs 2000, 60 (2), 379–411. 2. Land, S. R. JAMA 2007, 298 (9), 973–973. 3. Dadiboyena, S. Eur. J. Med. Chem. 2012, 51, 17–34. 4. Schmid, C. R.; Sluka, J. P.; Duke, K. M. Tetrahedron Lett. 1999, 40 (4), 675–678. 5. Bradley, D. A.; Godfrey, A. G.; Schmid, C. R. Tetrahedron Lett. 1999, 40 (28), 5155–5159. 6. Shinde, P. S.; Shinde, S. S.; Renge, A. S.; Patil, G. H.; Rode, A. B.; Pawar, R. R. Lett. Org. Chem. 2009, 6 (1), 8–10.7. Sach, N. W.; Richter, D. T.; Cripps, S.; Tran-Dube, M.; Zhu, H. C.; Huang, B. W.; Cui, J.; Sutton, S. C. Org. Lett. 2012, 14 (15), 3886–889. 8. Jones, C. D.; Jevnikar, M. G.; Pike, A. J.; Peters, M. K.; Black, L. J.; Thompson, A. R.; Falcone, J. F.; Clemens, J. A. J. Med. Chem. 1984, 27 (8), 1057–1066. 9. Grese, T. A.; Cho, S.; Finley, D. R.; Godfrey, A. G.; Jones, C. D.; Lugar, C. W.; Martin, M. J.; Matsumoto, K.; Pennington, L. D.; Winter, M. A.; Adrian, M. D.; Cole, H. W.; Magee, D. E.; Phillips, D. L.; Rowley, E. R.; Short, L. L.; Glasebrook, A. L.; Bryant, H. U. J. Med. Chem. 1997, 40 (2), 146–167. synChapter 2 – 1-Substituted PiperidinesAuthor links open overlay panelRubenVardanyan https://doi.org/10.1016/B978-0-12-805157-3.00002-8Piperidine-Based Drug DiscoveryHeterocyclic Drug Discovery2017, Pages 83-1011-Substituted Piperidines
Raloxifene (Evista) (1.3.4) is a second-generation selective estrogen receptor modulator that functions as an estrogen antagonist on breast and uterine tissues, and an estrogen agonist on bone. Raloxifene is an antiresorptive agent, a new representative of a class of drugs that prevent the loss of bone mass, i.e., used to treat osteoporosis and similar diseases in postmenopausal women and those postmenopausal women at increased risk of invasive breast cancer [41–53].
It was shown that raloxifene can have some affect on cognition, mental health, sleep, and sexual function in menopausal women [54]. Raloxifene was used also as an adjuvant treatment in postmenopausal women with schizophrenia [55].
The first reported synthesis of the raloxifene scaffold consists in Friedel-Crafts aroylation in 1,2-dichloroethane and using AlCl3 as a catalyst by coupling of 4-(2-(piperidin-1-yl)ethoxy)benzoyl chloride (2.3.15) with benzothiophene derivative (2.3.16) followed by alkaline hydrolysis of mesyl groups, which give the desired raloxifene (2.3.4) [56–58] (Scheme 2.9).
The key intermediate – 6-methoxy-2-(4-methoxyphenyl)benzo[b]thiophene (2.3.16) – was prepared by the cyclization-rearrangement of 1-(4-methoxyphenyl)-2-((3-methoxyphenyl)thio)ethan-1-one (2.3.20) induced by polyphosphoric acid (PPA). This rearrangement (Kost rearrangement [59]) is general for 3-(R-substituted)indoles, -benzofurans, and -benzothiophenes, which are converted to the corresponding 2-isomers by heating with PPA.
The synthesis started from thiophenol (2.3.18) and bromoketone (2.3.19), which were coupled in presence of KOH in ethanol/water solution. Obtained (2.3.20) was heated with PPA to give a mixture that is easily separable by crystallization isomeric 2-phenylbenzo[b]thiophenes (2.3.21) and (2.3.22), where preferable, isomer (2.3.22) predominates. Cleavage of the methoxy groups in (2.3.22) was done conveniently with pyridine hydrochloride to give (2.3.23), which was easily converted to mesylate (2.3.16) with methanesulfonyl chloride in pyridine and 4-dimethylaminopyridine as a catalyst (Scheme 2.10).
The second reagent—4-(2-(piperidin-1-yl)ethoxy)benzoyl chloride (2.3.15)—was prepared starting with 4-hydroxybenzoate (2.3.24), which with 1-(2-chloroethyl)piperidine (2.3.25) in anhydrous DMF, and K2CO3 or sodium hydride, gave methyl 4-(2-(piperidin-1-yl)ethoxy)benzoate (2.3.26) hydrolyzed in MeOH/water NaOH solution. The acid (2.3.26) was converted to its chloride (2.3.15) with SOCl2 in 1,2-dichloroethane and a catalytic amount of DMF (Scheme 2.11).
Another novel convenient synthesis of raloxifene (2.3.4) have been proposed [60]. According to this method anisaldehyde (2.3.28) was transformed to corresponding cyanohydrin (2.3.29) using a mixture of sodium cyanide ethanol containing triethylamine through which HCl gas was passed over 30 minutes at 5–10°C.
Gaseous HCl was added to the solution of prepared cyanohydrin (2.3.29) in ethanol at room temperature over 30 minutes in order to give p-methoxybenzaldehyde cyanohydrin iminoether hydrochloride (2.3.30). Then, hydrogen sulfide was bubbled into a solution of the methyl imidate (2.3.30) and triethylamine in methanol at 0°C to give α-(4-methoxy phenyl)-α-hydroxy-N,N dimethylthioacetamide (2.3.31).
To the obtained α-hydroxythioamide (2.3.31) dissolved-in-methylene chloride methanesulfonic acid was slowly added, which transformed the starting material to 2-N,N-dimethylamino-6-methoxy benzo[β]thiophene (2.3.32).
The obtained 2-dimethylaminobenzothiophene (2.3.32) and known 4-(2-piperidinoethoxy)-benzoyl chloride (2.3.15) were partially dissolved in chlorobenzene and the mixture was warmed in a 100–105°C to give 2-(4-methoxyphenyl)-6-methoxy-3-[4-(piperidinoethoxy)benzoyl]-benzo[β]thiophene (2.3.33). 4-Methoxyphenylmagnesium bromide (2.3.34) in THF was added to chilled to 0°C prepared compound (2.3.33) in THF, which gave 2-(4-methoxyphenyl)-6-methoxy-3-[4-(piperidinoethoxy)benzoyl] benzo[β] thiophene (2.3.35). To the prepared benzothiophene (2.3.35) suspended in chlorobenzene was added AlCl3, followed by the addition of n-propanethiol, and the mixture was heated at 35°C. After the workup with aqueous HCl, the desired raloxifene (2.3.4) was separated [60] (Scheme 2.12).
There exist plenty of modifications for these two approaches, as reviewed in [61,62].
^ Yang, Z. D.; Yu, J.; Zhang, Q. (August 2013). “Effects of raloxifene on cognition, mental health, sleep and sexual function in menopausal women: a systematic review of randomized controlled trials”. Maturitas. 75 (4): 341–348. doi:10.1016/j.maturitas.2013.05.010. ISSN1873-4111. PMID23764354.
^ Jump up to:abBritish national formulary : BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 736–737. ISBN9780857113382.
^ Ohta, Hiroaki; Hamaya, Etsuro; Taketsuna, Masanori; Sowa, Hideaki (January 2015). “Quality of life in Japanese women with postmenopausal osteoporosis treated with raloxifene and vitamin D: post hoc analysis of a postmarketing study”. Current Medical Research and Opinion. 31 (1): 85–94. doi:10.1185/03007995.2014.975339. ISSN1473-4877. PMID25299349. S2CID24671531.
^ Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V, Balaguer P (May 2006). “Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta”. Biochem. Pharmacol. 71 (10): 1459–69. doi:10.1016/j.bcp.2006.02.002. PMID16554039.
^ Greene, G. L.; Shiau, A. K.; Nettles, K. W. (2004). “A Structural Explanation for ERα/ERβ SERM Discrimination”. New Molecular Mechanisms of Estrogen Action and Their Impact on Future Perspectives in Estrogen Therapy (46): 33–45. doi:10.1007/978-3-662-05386-7_3. ISBN978-3-662-05388-1. PMID15248503.
^ Barkhem, Tomas; Carlsson, Bo; Nilsson, Yvonne; Enmark, Eva; Gustafsson, Jan-Åke; Nilsson, Stefan (1998). “Differential Response of Estrogen Receptor α and Estrogen Receptor β to Partial Estrogen Agonists/Antagonists”. Molecular Pharmacology. 54 (1): 105–112. doi:10.1124/mol.54.1.105. ISSN0026-895X. PMID9658195.
^ Jump up to:ab Uebelhart B, Herrmann F, Pavo I, Draper MW, Rizzoli R (September 2004). “Raloxifene treatment is associated with increased serum estradiol and decreased bone remodeling in healthy middle-aged men with low sex hormone levels”. J. Bone Miner. Res. 19 (9): 1518–24. doi:10.1359/JBMR.040503. PMID15312253. S2CID36104038.
^ Xu, Beibei; Lovre, Dragana; Mauvais-Jarvis, Franck (2016). “Effect of selective estrogen receptor modulators on metabolic homeostasis”. Biochimie. 124: 92–97. doi:10.1016/j.biochi.2015.06.018. ISSN0300-9084. PMID26133657. In healthy postmemopausal women, raloxifene treatment for one year prevented body weight gain and abdominal adiposity by promoting a shift from an android to gynoid fat distribution [46].
^ Francucci, C. M.; Pantaleo, D.; Iori, N.; Camilletti, A.; Massi, F.; Boscaro, M. (2014). “Effects of raloxifene on body fat distribution and lipid profile in healthy post-menopausal women”. Journal of Endocrinological Investigation. 28 (9): 623–631. doi:10.1007/BF03347261. ISSN0391-4097. PMID16218045. S2CID28467435. These results […] suggest, for the first time, that RLX promotes the shift from android to gynoid fat distribution, and prevents the uptrend of abdominal adiposity and body weight compared with untreated women.
^ Snyder KR, Sparano N, Malinowski JM (September 2000). “Raloxifene hydrochloride”. Am J Health Syst Pharm. 57 (18): 1669–75, quiz 1676–8. doi:10.1093/ajhp/57.18.1669. PMID11006795.
^ Provinciali N, Suen C, Dunn BK, DeCensi A (October 2016). “Raloxifene hydrochloride for breast cancer risk reduction in postmenopausal women”. Expert Rev Clin Pharmacol. 9 (10): 1263–1272. doi:10.1080/17512433.2016.1231575. PMID27583816. S2CID26047863.
^ Blum A, Hathaway L, Mincemoyer R, Schenke WH, Csako G, Waclawiw MA, Panza JA, Cannon RO (2000). “Hormonal, lipoprotein, and vascular effects of the selective estrogen receptor modulator raloxifene in hypercholesterolemic men”. Am. J. Cardiol. 85 (12): 1491–4, A7. doi:10.1016/s0002-9149(00)00802-x. PMID10856400.
^ Doran PM, Riggs BL, Atkinson EJ, Khosla S (2001). “Effects of raloxifene, a selective estrogen receptor modulator, on bone turnover markers and serum sex steroid and lipid levels in elderly men”. J. Bone Miner. Res. 16 (11): 2118–25. doi:10.1359/jbmr.2001.16.11.2118. PMID11697809. S2CID28216610.
^ Jump up to:ab Ho TH, Nunez-Nateras R, Hou YX, Bryce AH, Northfelt DW, Dueck AC, Wong B, Stanton ML, Joseph RW, Castle EP (2017). “A Study of Combination Bicalutamide and Raloxifene for Patients With Castration-Resistant Prostate Cancer”. Clin Genitourin Cancer. 15 (2): 196–202.e1. doi:10.1016/j.clgc.2016.08.026. PMID27771244. S2CID19043552.
^ Khodaie-Ardakani MR, Khosravi M, Zarinfard R, Nejati S, Mohsenian A, Tabrizi M, Akhondzadeh S (2015). “A Placebo-Controlled Study of Raloxifene Added to Risperidone in Men with Chronic Schizophrenia”. Acta Med Iran. 53 (6): 337–45. PMID26069170.
^ Jump up to:abc Wang Q, Dong X, Wang Y, Li X (2017). “Raloxifene as an adjunctive treatment for postmenopausal women with schizophrenia: a meta-analysis of randomized controlled trials”. Arch Womens Ment Health. 21 (1): 31–41. doi:10.1007/s00737-017-0773-2. PMID28849318. S2CID4524617.
^ Carneiro AL, de Cassia de Maio Dardes R, Haidar MA (July 2012). “Estrogens plus raloxifene on endometrial safety and menopausal symptoms–semisystematic review”. Menopause. 19 (7): 830–4. doi:10.1097/gme.0b013e31824a74ce. PMID22549172. S2CID196380398.
^ Leung KC, Leung AC (2017). “Gynecomastia in Infants, Children, and Adolescents”. Recent Pat Endocr Metab Immune Drug Discov. 10 (2): 127–137. doi:10.2174/1872214811666170301124033. PMID28260521.
^ Lawrence SE, Faught KA, Vethamuthu J, Lawson ML (July 2004). “Beneficial effects of raloxifene and tamoxifen in the treatment of pubertal gynecomastia”. J. Pediatr. 145 (1): 71–6. doi:10.1016/j.jpeds.2004.03.057. PMID15238910.
Heringa M (2003). “Review on raloxifene: profile of a selective estrogen receptor modulator”. Int J Clin Pharmacol Ther. 41 (8): 331–45. doi:10.5414/cpp41331. PMID12940590.
Sporn MB, Dowsett SA, Mershon J, Bryant HU (2004). “Role of raloxifene in breast cancer prevention in postmenopausal women: clinical evidence and potential mechanisms of action”. Clin Ther. 26 (6): 830–40. doi:10.1016/s0149-2918(04)90127-0. PMID15262454.
Yang ZD, Yu J, Zhang Q (2013). “Effects of raloxifene on cognition, mental health, sleep and sexual function in menopausal women: a systematic review of randomized controlled trials”. Maturitas. 75 (4): 341–8. doi:10.1016/j.maturitas.2013.05.010. PMID23764354.
External links
“Raloxifene”. Drug Information Portal. U.S. National Library of Medicine.
Molecular Formula: C13H16N2O2Molecular Weight: 232.28Percent Composition: C 67.22%, H 6.94%, N 12.06%, O 13.78%
Literature References: A hormone of the pineal gland, also produced by extra-pineal tissues, that lightens skin color in amphibians by reversing the darkening effect of MSH, q.v. Melatonin has been postulated as the mediator of photic-induced antigonadotrophic activity in photoperiodic mammals and has also been shown to be involved in thermoregulation in some ectotherms and in affecting locomotor activity rhythms in sparrows. Isoln from the pineal glands of beef cattle: Lerner et al.,J. Am. Chem. Soc.80, 2587 (1958); Wurtman et al.,Science141, 277 (1963). Structure: Lerner et al.,J. Am. Chem. Soc.81, 6084 (1959). Crystal and molecular structure: A. Wakahara, Chem. Lett.1972, 1139. Synthesis from 5-methoxyindole as starting material by two different routes: Szmuszkovicz et al.,J. Org. Chem.25, 857 (1960). Biochemical role of melatonin: Chem. Eng. News45, 40 (May 1, 1967). Pharmacological studies: Barchas et al.,Nature214, 919 (1967). Identification of antigonadal action sites in mouse brain: J. D. Glass, G. R. Lynch, Science214, 821 (1981). Binding studies in human hypothalamus: S. M. Reppert et al.,Science242, 78 (1988). Efficacy in control of estrus in red deer: G. W. Asher, Anim. Reprod. Sci.22, 145 (1990). Reviews: M. K. Vaughn, Int. J. Rev. Physiol.24, 41-95 (1981); D. C.Klein et al.,Life Sci.28, 1975-1986 (1981). Book: Advan. Biosci.vol. 29, N. Birau, W. Schlott, Eds. (Pergamon Press, New York, 1981) 420 pp. Review of etiological role in clinical disease: A. Miles, D. Philbrick, Crit. Rev. Clin. Lab. Sci.25, 231-253 (1987); in psychiatric disorders: eidem,Biol. Psychiatry23, 405-425 (1988).Properties: Pale yellow leaflets from benzene, mp 116-118°. uv max: 223, 278 nm (e 27550, 6300).Melting point: mp 116-118°Absorption maximum: uv max: 223, 278 nm (e 27550, 6300)Therap-Cat-Vet: Control of estrus.
Melatonin is a hormone primarily released by the pineal gland that regulates the sleep–wake cycle.[3][4] As a dietary supplement, it is often used for the short-term treatment of insomnia, such as from jet lag or shift work, and is typically taken by mouth.[5][6][7] Evidence of its benefit for this use, however, is not strong.[8] A 2017 review found that sleep onset occurred six minutes faster with use, but found no change in total time asleep.[6] The melatonin receptor agonist medication ramelteon may work as well as melatonin supplements,[6] at greater cost but with different adverse effects, for some sleep conditions.[9]
Melatonin was discovered in 1958.[3] It is sold over the counter in Canada and the United States;[10][13] in the United Kingdom, it is a prescription-only medication.[7] It is not approved by the US Food and Drug Administration (FDA) for any medical use.[10] In Australia and the European Union, it is indicated for difficulty sleeping in people over the age of 54.[20][11] In the European Union, it is indicated for the treatment of insomnia in children and adolescents.[12] It was approved for medical use in the European Union in 2007.[11]
Chemical Synthesis of Melatonin The methods for the chemical synthesis of melatonin are generally not so complicated and do not involve more than three steps of conversion. Three synthesis reactions of melatonin from primary literatures are shown below;
Reaction 1
In 1958 melatonin was first isolated and characterised by A.B.Lerner. It was know as one of a substituted 5-hydroxyindole derivative in the pineal gland that could lighten pigment cells. It had not been know to exist in biological tissue although it had been isolated as a urinary excretion product in rats after administration of 5-hydroxytryptamine. Melatonin or N-acetyl-5-methoxytryptamine (40 mg) was prepared by reducing 100 mg of 5-methoxyindole-3-acetonitrile with 160 mg of sodium and 2 ml of ethanol. Then the product was acetylated with 4 ml of both glacial acetic acid and acetic anhydride at 100 oC for 1 minute. Purification was achieved by countercerrent distribution and silicic acid chromatography.
Reaction 2
5-Methoxytryptamine hydrochloride (1g, 4.75 mmole) was dissolved in pyridine (10 ml) and acetic anhydride (10 ml) and kept overnight at 20 oC. The solution was poured onto iced, neutralised with dilute hydrochloric acid and extracted with chloroform (2×25 ml). The combined extracts were washed with water, dried in MgSO4 and evaporated to afford a liquid of N,N diacetyltryptamine derivative. The liquid was then poured into water (50 ml) and extracted with chlroform (2×25 ml). The combined organic layers were washed with water (25 ml), dried in MgSO4 and evaporated to dryness. The residual solid crystallised from benzene to afford melatonin 819 mg, 80% yield.
Reaction 3
The more reactive indoles (1a-1d) were alkylated at the 3 position by reaction with nitroethene generated in situ by thermolysis of nitroethyl acetate. The nitroethyl acetate used for this purpose was prepared by acetylation of nitroethanol with acetic anhydride using NaOAc as a catalyst. These conditions constitute a substantial improvement of the overal yield of the reation. Reduction of the nitroethylated indoles (2a-d) by hydrogenation over PtO2, followed by acetylation fo the resluting tryptamines with acetic anhydride-pyridine completed the synthesis of melatonin and its derivatives (4a-d).
Biological Synthesis and Metabolism of Melatonin
The biosynthesis of melatonin (Fig.1) is initiated by the uptake of the essential amino acid tryptophan into pineal parenchymal cells. Tryptophan is the least abundant of essential amino acids in normal diets. It is converted to another amino acid, 5-hydroxytryptophan, through the action of the enzyme tryptopahn hydroxylase and then to 5-hydroxytryptamine (serotonin) by the enzyme aromatic amino acid decarboxylase. Serotonin concentrations are higher in the pineal than in any other organ or in any brain region. They exhibit a striking diurnal rhythm remaining at a maximum level during the daylight hours and falling by more than 80% soon after the onset of darkness as the serotonin is converted to melatonin, 5-hydroxytryptophol and other methoxyindoles. Serotonin’s conversion to melatonin involves two enzymes that are characteristic of the pineal : SNAT (serotonin-N-acetyltransferase) which converts the serotonin to N-acetylserotonin, and HIOMT (hydroxyindole-O-methyltrasferase) which trasfers a methyl group from S-adenosylmethionine to the 5-hydroxyl of the N-acetylserotonin. The activities of both enzymes rise soon after the onset of darkness because of the enhanced release of norepinephrine from sympathetic neurons terminating on the pineal parenchymal cells. Another portion of the serotonin liberated from pineal cells after the onset of darkness is deaminated by the enzyme monoamine oxidase (MAO) and then either oxidized to form 5-hydroxyindole acetic acid or reduced to form 5-hydroxytryptophol (Fig.1). Both of these compounds are also substrates for HIOMT and can thus be converted in the pineal to 5-methoxyindole acetic acid 5-methoxytryptophol (Fig.1). The level of this latter indole, like that of melatonin, rises markedly in the pineal with the onset of darkness. Since 5-methoxytryptophol synthesis does not require the acetylation of serotonin, the nocturnal increase in pineal SNAT activity cannot be the trigger that causes pineal methoxyindole levels to rise. More likely, a single unexplained process- the intraparenchymal release of stored pineal serotonin, which then becomes accessible to both SNAT and MAO. This process ultimately controls the rates at which all three major pineal methoxyindoles are synthesized and generates the nocturnal increases in pineal melatonin and 5-methoxytryptophol. The proportion of available serotonin acetylated at any particular time of day or night depends on the relative activities of pineal SNAT and MAO at that time. The rates of methylation of all three 5-hydroxyindoles formed from pinela serotonin depends on HIOMT activity.Fig.1 Biosynthesis of pineal methoxyindoles from serotonin
Serotonin may be either acetylated to form N-acetylserotonin through the action of the enzyme serotonin-N-acetyltransferase (SNAT), or oxidatively deaminated by monoamine oxidase (MAO) to yield an unstable aldehyde. This compound is then either oxidized to 5-hydroxyindole acetic acid by the enzyme aldehyde dehydrogenase (ADH), or reduced to from 5-hydroxytryptophol by aldehyde reductase (AR). Each of these 5-hydroxyindole derivatives of serotonin is a substrate for hydroxyindole-O-methyltrasferase (HIMOT). The enzymatic trasfer of a methyl group from S-adenosylmethionine to these hydroxyindoles yields melatonin (5-hydroxy-N-acetyltryptamine), 5-methoxyindole acetic acid and 5-methoxytryptophol respectively. Pineal serotonin is synthesized from the essential amino acid tryptophan by 5-hydroxylation folloed by decarboxylation. The first step in ths enzymic sequence is catalysed by tryptophan hydroxylase. The second step is catalysed by aromatic L-amino acid decarboxylase.
Medical uses
In the European Union it is indicated for the treatment of insomnia in children and adolescents aged 2–18 with autism spectrum disorder (ASD) and / or Smith–Magenis syndrome, where sleep hygiene measures have been insufficient[12] and for monotherapy for the short-term treatment of primary insomnia characterized by poor quality of sleep in people who are aged 55 or over.[11]
Sleep disorders
Positions on the benefits of melatonin for insomnia are mixed.[8] An Agency for Healthcare Research and Quality (AHRQ) review from 2015 stated that evidence of benefit in the general population was unclear.[8] A review from 2017, found a modest effect on time until onset of sleep.[3] Another review from 2017 put this decrease at six minutes to sleep onset but found no difference in total sleep time.[6] Melatonin may also be useful in delayed sleep phase syndrome.[3] Melatonin appears to work as well as ramelteon but costs less.[6]
A 2020 Cochrane review found no evidence that melatonin helped sleep problems in people with moderate to severe dementia due to Alzheimer’s disease.[26] A 2019 review found that while melatonin may improve sleep in minimal cognitive impairment, after the onset of Alzheimer’s it has little to no effect.[27] Melatonin may, however, help with sundowning.[28]
Jet lag and shift work
Melatonin is known to reduce jet lag, especially in eastward travel. If the time it is taken is not correct, however, it can instead delay adaption.[29]
Melatonin appears to have limited use against the sleep problems of people who work shift work.[30] Tentative evidence suggests that it increases the length of time people are able to sleep.[30]
Adverse effects
Melatonin appears to cause very few side effects as tested in the short term, up to three months, at low doses.[clarification needed] Two systematic reviews found no adverse effects of exogenous melatonin in several clinical trials and comparative trials found the adverse effects headaches, dizziness, nausea, and drowsiness were reported about equally for both melatonin and placebo.[31][32] Prolonged-release melatonin is safe with long-term use of up to 12 months.[33] Although not recommended for long term use beyond this, low-dose melatonin is generally safer, and a better alternative, than many prescription and over the counter sleep aids if a sleeping medication must be used for an extended period of time. Low-doses of melatonin are usually sufficient to produce a hypnotic effect in most people. Higher doses do not appear to result in a stronger effect, but instead appear to cause drowsiness for a longer period of time.[34]
In those taking warfarin, some evidence suggests there may exist a potentiating drug interaction, increasing the anticoagulant effect of warfarin and the risk of bleeding.[41]
Functions
When eyes receive light from the sun, the pineal gland’s production of melatonin is inhibited and the hormones produced keep the human awake. When the eyes do not receive light, melatonin is produced in the pineal gland and the human becomes tired.
Circadian rhythm
In animals, melatonin plays an important role in the regulation of sleep–wake cycles.[42] Human infants’ melatonin levels become regular in about the third month after birth, with the highest levels measured between midnight and 8:00 am.[43] Human melatonin production decreases as a person ages.[44] Also, as children become teenagers, the nightly schedule of melatonin release is delayed, leading to later sleeping and waking times.[45]
Antioxidant
Melatonin was first reported as a potent antioxidant and free radical scavenger in 1993.[46] In vitro, melatonin acts as a direct scavenger of oxygen radicals and reactive nitrogen species including OH•, O2−•, and NO•.[47][48] In plants, melatonin works with other antioxidants to improve the overall effectiveness of each antioxidant.[48] Melatonin has been proven to be twice as active as vitamin E, believed to be the most effective lipophilic antioxidant.[49] Via signal transduction through melatonin receptors, melatonin promotes the expression of antioxidant enzymes such as superoxide dismutase, glutathione peroxidase, glutathione reductase, and catalase.[50][51]
Melatonin occurs at high concentrations within mitochondrial fluid which greatly exceed the plasma concentration of melatonin.[52][53][54] Due to its capacity for free radical scavenging, indirect effects on the expression of antioxidant enzymes, and its significant concentrations within mitochondria, a number of authors have indicated that melatonin has an important physiological function as a mitochondrial antioxidant.[50][52][53][54][55]
The melatonin metabolites produced via the reaction of melatonin with reactive oxygen species or reactive nitrogen species also react with and reduce free radicals.[51][55] Melatonin metabolites generated from redox reactions include cyclic 3-hydroxymelatonin, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), and N1-acetyl-5-methoxykynuramine (AMK).[51][55]
Immune system
While it is known that melatonin interacts with the immune system,[56][57] the details of those interactions are unclear. An antiinflammatory effect seems to be the most relevant. There have been few trials designed to judge the effectiveness of melatonin in disease treatment. Most existing data are based on small, incomplete trials. Any positive immunological effect is thought to be the result of melatonin acting on high-affinity receptors (MT1 and MT2) expressed in immunocompetent cells. In preclinical studies, melatonin may enhance cytokine production,[58] and by doing this, counteract acquired immunodeficiences. Some studies also suggest that melatonin might be useful fighting infectious disease[59] including viral, such as HIV, and bacterial infections, and potentially in the treatment of cancer.
In bacteria, protists, fungi, and plants, melatonin is synthesized indirectly with tryptophan as an intermediate product of the shikimate pathway. In these cells, synthesis starts with D-erythrose 4-phosphate and phosphoenolpyruvate, and in photosynthetic cells with carbon dioxide. The rest of the synthesising reactions are similar, but with slight variations in the last two enzymes.[62][63]
It has been hypothesized that melatonin is made in the mitochondria and chloroplasts.[64]
Mechanism
Mechanism of melatonin biosynthesis
In order to hydroxylate L-tryptophan, the cofactor tetrahydrobiopterin (THB) must first react with oxygen and the active site iron of tryptophan hydroxylase. This mechanism is not well understood, but two mechanisms have been proposed:
1. A slow transfer of one electron from the THB to O2 could produce a superoxide which could recombine with the THB radical to give 4a-peroxypterin. 4a-peroxypterin could then react with the active site iron (II) to form an iron-peroxypterin intermediate or directly transfer an oxygen atom to the iron.
2. O2 could react with the active site iron (II) first, producing iron (III) superoxide which could then react with the THB to form an iron-peroxypterin intermediate.
Iron (IV) oxide from the iron-peroxypterin intermediate is selectively attacked by a double bond to give a carbocation at the C5 position of the indole ring. A 1,2-shift of the hydrogen and then a loss of one of the two hydrogen atoms on C5 reestablishes aromaticity to furnish 5-hydroxy-L-tryptophan.[65]
A decarboxylase with cofactor pyridoxal phosphate (PLP) removes CO2 from 5-hydroxy-L-tryptophan to produce 5-hydroxytryptamine.[66] PLP forms an imine with the amino acid derivative. The amine on the pyridine is protonated and acts as an electron sink, enabling the breaking of the C-C bond and releasing CO2. Protonation of the amine from tryptophan restores the aromaticity of the pyridine ring and then imine is hydrolyzed to produce 5-hydroxytryptamine and PLP.[67]
It has been proposed that histidine residue His122 of serotonin N-acetyl transferase is the catalytic residue that deprotonates the primary amine of 5-hydroxytryptamine, which allows the lone pair on the amine to attack acetyl-CoA, forming a tetrahedral intermediate. The thiol from coenzyme A serves as a good leaving group when attacked by a general base to give N-acetylserotonin.[68]
N-acetylserotonin is methylated at the hydroxyl position by S-adenosyl methionine (SAM) to produce S-adenosyl homocysteine (SAH) and melatonin.[67][69]
Regulation
In vertebrates, melatonin secretion is regulated by activation of the beta-1 adrenergic receptor by norepinephrine.[70] Norepinephrine elevates the intracellular cAMP concentration via beta-adrenergic receptors and activates the cAMP-dependent protein kinase A (PKA). PKA phosphorylates the penultimate enzyme, the arylalkylamine N-acetyltransferase (AANAT). On exposure to (day)light, noradrenergic stimulation stops and the protein is immediately destroyed by proteasomalproteolysis.[71] Production of melatonin is again started in the evening at the point called the dim-light melatonin onset.
Blue light, principally around 460–480 nm, suppresses melatonin biosynthesis,[72] proportional to the light intensity and length of exposure. Until recent history, humans in temperate climates were exposed to few hours of (blue) daylight in the winter; their fires gave predominantly yellow light.[citation needed] The incandescent light bulb widely used in the 20th century produced relatively little blue light.[73] Light containing only wavelengths greater than 530 nm does not suppress melatonin in bright-light conditions.[74] Wearing glasses that block blue light in the hours before bedtime may decrease melatonin loss. Use of blue-blocking goggles the last hours before bedtime has also been advised for people who need to adjust to an earlier bedtime, as melatonin promotes sleepiness.[75]
When used several hours before sleep according to the phase response curve for melatonin in humans, small amounts (0.3 mg[77]) of melatonin shift the circadian clock earlier, thus promoting earlier sleep onset and morning awakening.[78] Melatonin is rapidly absorbed and distributed, reaching peak plasma concentrations after 60 minutes of administration, and is then eliminated.[61] Melatonin has a half life of 35–50 minutes.[79] In humans, 90% of orally administered exogenous melatonin is cleared in a single passage through the liver, a small amount is excreted in urine, and a small amount is found in saliva.[5] The bioavalibility of melatonin is between 10 and 50%.[61]
Melatonin is metabolized in the liver by cytochrome P450 enzyme CYP1A2 to 6-hydroxymelatonin. Metabolites are conjugated with sulfuric acid or glucuronic acid for excretion in the urine. 5% of melatonin is excreted in the urine as the unchanged drug.[61]
Some of the metabolites formed via the reaction of melatonin with a free radical include cyclic 3-hydroxymelatonin, N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK), and N1-acetyl-5-methoxykynuramine (AMK).[51][55]
In 1958, dermatology professor Aaron B. Lerner and colleagues at Yale University, in the hope that a substance from the pineal might be useful in treating skin diseases, isolated the hormone from bovine pineal gland extracts and named it melatonin.[85] In the mid-70s Lynch et al. demonstrated that the production of melatonin exhibits a circadian rhythm in human pineal glands.[86]
The discovery that melatonin is an antioxidant was made in 1993.[87] The first patent for its use as a low-dose sleep aid was granted to Richard Wurtman at MIT in 1995.[88] Around the same time, the hormone got a lot of press as a possible treatment for many illnesses.[89]The New England Journal of Medicine editorialized in 2000: “With these recent careful and precise observations in blind persons, the true potential of melatonin is becoming evident, and the importance of the timing of treatment is becoming clear.”[90]
It was approved for medical use in the European Union in 2007.[11]
Other animals
In vertebrates, melatonin is produced in darkness, thus usually at night, by the pineal gland, a small endocrine gland[91] located in the center of the brain but outside the blood–brain barrier. Light/dark information reaches the suprachiasmatic nuclei from retinal photosensitive ganglion cells of the eyes[92][93] rather than the melatonin signal (as was once postulated). Known as “the hormone of darkness”, the onset of melatonin at dusk promotes activity in nocturnal (night-active) animals and sleep in diurnal ones including humans.
Many animals use the variation in duration of melatonin production each day as a seasonal clock.[94] In animals including humans,[95] the profile of melatonin synthesis and secretion is affected by the variable duration of night in summer as compared to winter. The change in duration of secretion thus serves as a biological signal for the organization of daylength-dependent (photoperiodic) seasonal functions such as reproduction, behavior, coat growth, and camouflage coloring in seasonal animals.[95] In seasonal breeders that do not have long gestation periods and that mate during longer daylight hours, the melatonin signal controls the seasonal variation in their sexual physiology, and similar physiological effects can be induced by exogenous melatonin in animals including mynah birds[96] and hamsters.[97] Melatonin can suppress libido by inhibiting secretion of luteinizing hormone and follicle-stimulating hormone from the anterior pituitary gland, especially in mammals that have a breeding season when daylight hours are long. The reproduction of long-day breeders is repressed by melatonin and the reproduction of short-day breeders is stimulated by melatonin.
During the night, melatonin regulates leptin, lowering its levels.
Cetaceans have lost all the genes for melatonin synthesis as well as those for melatonin receptors.[98] This is thought to be related to their unihemispheric sleep pattern (one brain hemisphere at a time). Similar trends have been found in sirenians.[98]
Plants
Until its identification in plants in 1987, melatonin was for decades thought to be primarily an animal neurohormone. When melatonin was identified in coffee extracts in the 1970s, it was believed to be a byproduct of the extraction process. Subsequently, however, melatonin has been found in all plants that have been investigated. It is present in all the different parts of plants, including leaves, stems, roots, fruits, and seeds, in varying proportions.[19][99] Melatonin concentrations differ not only among plant species, but also between varieties of the same species depending on the agronomic growing conditions, varying from picograms to several micrograms per gram.[63][100] Notably high melatonin concentrations have been measured in popular beverages such as coffee, tea, wine, and beer, and crops including corn, rice, wheat, barley, and oats.[19] In some common foods and beverages, including coffee[19] and walnuts,[101] the concentration of melatonin has been estimated or measured to be sufficiently high to raise the blood level of melatonin above daytime baseline values.
Although a role for melatonin as a plant hormone has not been clearly established, its involvement in processes such as growth and photosynthesis is well established. Only limited evidence of endogenous circadian rhythms in melatonin levels has been demonstrated in some plant species and no membrane-bound receptors analogous to those known in animals have been described. Rather, melatonin performs important roles in plants as a growth regulator, as well as environmental stress protector. It is synthesized in plants when they are exposed to both biological stresses, for example, fungal infection, and nonbiological stresses such as extremes of temperature, toxins, increased soil salinity, drought, etc.[63][102][103]
Occurrence
Dietary supplement
Melatonin is categorized by the US Food and Drug Administration (FDA) as a dietary supplement, and is sold over-the-counter in both the US and Canada.[5] FDA regulations applying to medications are not applicable to melatonin,[15] though the FDA has found false claims that it cures cancer.[104] As melatonin may cause harm in combination with certain medications or in the case of certain disorders, a doctor or pharmacist should be consulted before making a decision to take melatonin.[29] In many countries, melatonin is recognized as a neurohormone and it cannot be sold over-the-counter.[105]
Food products
Naturally-occurring melatonin has been reported in foods including tart cherries to about 0.17–13.46 ng/g,[106] bananas and grapes, rice and cereals, herbs, plums,[107] olive oil, wine[108] and beer. When birds ingest melatonin-rich plant feed, such as rice, the melatonin binds to melatonin receptors in their brains.[109] When humans consume foods rich in melatonin, such as banana, pineapple, and orange, the blood levels of melatonin increase significantly.[110]
Beverages and snacks containing melatonin were being sold in grocery stores, convenience stores, and clubs in May 2011.[111] The FDA considered whether these food products could continue to be sold with the label “dietary supplements”. On 13 January 2010, it issued a Warning Letter to Innovative Beverage, creators of several beverages marketed as drinks, stating that melatonin, while legal as a dietary supplement, was not approved as a food additive.[112] A different company selling a melatonin-containing beverage received a warning letter in 2015.[113]
Commercial availability
Immediate-release melatonin is not tightly regulated in countries where it is available as an over-the-counter medication. It is available in doses from less than half a milligram to 5 mg or more. Immediate-release formulations cause blood levels of melatonin to reach their peak in about an hour. The hormone may be administered orally, as capsules, gummies, tablets, or liquids. It is also available for use sublingually, or as transdermal patches.[medical citation needed]
Formerly, melatonin was derived from animal pineal tissue, such as bovine. It is now synthetic, which limits the risk of contamination or the means of transmitting infectious material.[15][114]
Melatonin is the most popular over-the-counter sleep remedy in the US, resulting in sales in excess of US$400 million during 2017.[115]
Research
A bottle of melatonin tablets. Melatonin is available in timed-release and in liquid forms.
Various uses and effects of melatonin have been studied. A 2015 review of studies of melatonin in tinnitus found the quality of evidence low, but not entirely without promise.[116]
Headaches
Tentative evidence shows melatonin may help reduce some types of headaches including cluster and hypnic headaches.[117][118]
Cancer
A 2013 review by the National Cancer Institutes found evidence for use to be inconclusive.[119] A 2005 review of unblinded clinical trials found a reduced rate of death, but that blinded and independently conducted randomized controlled trials are needed.[120]
Protection from radiation
Both animal[121] and human[122][123][124] studies have shown melatonin to protect against radiation-induced cellular damage. Melatonin and its metabolites protect organisms from oxidative stress by scavenging reactive oxygen species which are generated during exposure.[125] Nearly 70% of biological damage caused by ionizing radiation is estimated to be attributable to the creation of free radicals, especially the hydroxyl radical that attacks DNA, proteins, and cellular membranes. Melatonin has been described as a broadly protective, readily available, and orally self-administered antioxidant that is without known, major side effects.[126]
Epilepsy
A 2016 review found no beneficial role of melatonin in reducing seizure frequency or improving quality of life in people with epilepsy.[127]
Secondary dysmenorrhoea
A 2016 review suggested no strong evidence of melatonin compared to placebo for dysmenorrhoea secondary to endometriosis.[128]
Delirium
A 2016 review suggested no clear evidence of melatonin to reduce the incidence of delirium.[129]
Gastroesophageal reflux disease
A 2011 review said melatonin is effective in relieving epigastric pain and heartburn.[130]
Psychiatry
Melatonin might improve sleep in people with autism.[131] Children with autism have abnormal melatonin pathways and below-average physiological levels of melatonin.[132][133] Melatonin supplementation has been shown to improve sleep duration, sleep onset latency, and night-time awakenings.[132][134][135] However, many studies on melatonin and autism rely on self-reported levels of improvement and more rigorous research is needed.
While the packaging of melatonin often warns against use in people under 18 years of age, studies suggest that melatonin is an efficacious and safe treatment for insomnia in people with ADHD, including children. However, larger and longer studies are needed to establish long-term safety and optimal dosing.[136]
Melatonin in comparison to placebo is effective for reducing preoperative anxiety in adults when given as premedication. It may be just as effective as standard treatment with midazolam in reducing preoperative anxiety. Melatonin may also reduce postoperative anxiety (measured 6 hours after surgery) when compared to placebo.[137]
Some supplemental melatonin users report an increase in vivid dreaming. Extremely high doses of melatonin increased REM sleep time and dream activity in people both with and without narcolepsy.[138] Some evidence supports an antidepressant effect.[139]
^ Jump up to:abc Buscemi N, Vandermeer B, Pandya R, Hooton N, Tjosvold L, Hartling L, et al. (November 2004). “Melatonin for treatment of sleep disorders” (PDF). Evidence Report/Technology Assessment No. 108. (Prepared by the University of Alberta Evidence-based Practice Center, Under Contract No. 290-02-0023.) AHRQ Publication No. 05-E002-2. Rockville, MD: Agency for Healthcare Research and Quality. Agency for Healthcare Research and Quality (AHRQ), US Department of Health and Human Services (108): 1–7. doi:10.1037/e439412005-001. PMC4781368. PMID15635761. Retrieved 5 June2013.
^ Jump up to:abcdef Matheson E, Hainer BL (July 2017). “Insomnia: Pharmacologic Therapy”. American Family Physician. 96 (1): 29–35. PMID28671376.
^ Jump up to:abc Brasure M, MacDonald R, Fuchs E, Olson CM, Carlyle M, Diem S, et al. (2015). “Management of Insomnia Disorder[Internet]”. AHRQ Comparative Effectiveness Reviews. 15 (16): EHC027–EF. PMID26844312. Evidence for benzodiazepine hypnotics, melatonin agonists in the general adult population, and most pharmacologic interventions in older adults was generally insufficient
^ Adams, Katie S. (2014). “Melatonin agonists in the management of sleep disorders: A focus on ramelteon and tasimelteon”. Mental Health Clinician. 4 (2): 59–64. doi:10.9740/mhc.n190087. Retrieved 25 October 2020. However, the clinical relevance of this objective and therefore the author’s conclusion that these results support the potential use of ramelteon in circadian rhythm sleep disorders is questionable. … It is unclear whether Takeda Pharmaceuticals will pursue FDA indications for ramelteon for circadian rhythm disorders given these results.
^ Boutin JA, Audinot V, Ferry G, Delagrange P (August 2005). “Molecular tools to study melatonin pathways and actions”. Trends in Pharmacological Sciences. 26 (8): 412–9. doi:10.1016/j.tips.2005.06.006. PMID15992934.
^ Hardeland R (July 2005). “Antioxidative protection by melatonin: multiplicity of mechanisms from radical detoxification to radical avoidance”. Endocrine. 27 (2): 119–30. doi:10.1385/ENDO:27:2:119. PMID16217125.
^“Australian Public Assessment Report for Melatonin” (PDF). Australian Government Department of Health and Ageing Therapeutic Goods Administration. January 2011. pp. 2, 4. Retrieved 9 January 2019. Monotherapy for the short term treatment of primary insomnia characterised by poor quality of sleep in patients who are aged 55 or over.
^ Gao C, Scullin MK, Bliwise DL (2019). “Mild Cognitive Impairment and Dementia”. In Savard J, Ouellet MC (eds.). Handbook of Sleep Disorders in Medical Conditions. Academic Press. pp. 253–276. doi:10.1016/b978-0-12-813014-8.00011-1. ISBN978-0-12-813014-8.
^ Lyseng-Williamson KA (November 2012). “Melatonin prolonged release: in the treatment of insomnia in patients aged ≥55 years”. Drugs & Aging. 29 (11): 911–23. doi:10.1007/s40266-012-0018-z. PMID23044640. S2CID1403262.
^ Database of Abstracts of Reviews of Effects (DARE): Quality-assessed Reviews [Internet]. York (UK): Centre for Reviews and Dissemination (UK); 1995. Optimal dosages for melatonin supplementation therapy in older adults: a systematic review of current literature. 2014.
^ Morera AL, Henry M, de La Varga M (2001). “[Safety in melatonin use]” [Safety in melatonin use]. Actas Espanolas de Psiquiatria (in Spanish). 29 (5): 334–7. PMID11602091.
^ Ardura J, Gutierrez R, Andres J, Agapito T (2003). “Emergence and evolution of the circadian rhythm of melatonin in children”. Hormone Research. 59 (2): 66–72. doi:10.1159/000068571. PMID12589109. S2CID41937922.
^ Poeggeler B, Saarela S, Reiter RJ, Tan DX, Chen LD, Manchester LC, Barlow-Walden LR (November 1994). “Melatonin—a highly potent endogenous radical scavenger and electron donor: new aspects of the oxidation chemistry of this indole accessed in vitro”. Annals of the New York Academy of Sciences. 738 (1): 419–20. Bibcode:1994NYASA.738..419P. doi:10.1111/j.1749-6632.1994.tb21831.x. PMID7832450. S2CID36383425.
^ Pieri C, Marra M, Moroni F, Recchioni R, Marcheselli F (1994). “Melatonin: a peroxyl radical scavenger more effective than vitamin E”. Life Sciences. 55 (15): PL271-6. doi:10.1016/0024-3205(94)00666-0. PMID7934611.
^ Jump up to:abcdefgh Jockers R, Delagrange P, Dubocovich ML, Markus RP, Renault N, Tosini G, et al. (September 2016). “Update on melatonin receptors: IUPHAR Review 20”. British Journal of Pharmacology. 173 (18): 2702–25. doi:10.1111/bph.13536. PMC4995287. PMID27314810. Hence, one melatonin molecule and its associated metabolites could scavenge a large number of reactive species, and thus, the overall antioxidant capacity of melatonin is believed to be greater than that of other well‐known antioxidants, such as vitamin C and vitamin E, under in vitro or in vivo conditions (Gitto et al., 2001; Sharma and Haldar, 2006; Ortiz et al., 2013).
^ Jump up to:abc Reiter RJ, Rosales-Corral S, Tan DX, Jou MJ, Galano A, Xu B (November 2017). “Melatonin as a mitochondria-targeted antioxidant: one of evolution’s best ideas”. Cellular and Molecular Life Sciences. 74 (21): 3863–3881. doi:10.1007/s00018-017-2609-7. PMID28864909. S2CID23820389. melatonin is specifically targeted to the mitochondria where it seems to function as an apex antioxidant … The measurement of the subcellular distribution of melatonin has shown that the concentration of this indole in the mitochondria greatly exceeds that in the blood.
^ Jump up to:abc Reiter RJ, Mayo JC, Tan DX, Sainz RM, Alatorre-Jimenez M, Qin L (October 2016). “Melatonin as an antioxidant: under promises but over delivers”. Journal of Pineal Research. 61 (3): 253–78. doi:10.1111/jpi.12360. PMID27500468. S2CID35435683. There is credible evidence to suggest that melatonin should be classified as a mitochondria-targeted antioxidant.
^ Jump up to:abc Manchester LC, Coto-Montes A, Boga JA, Andersen LP, Zhou Z, Galano A, et al. (November 2015). “Melatonin: an ancient molecule that makes oxygen metabolically tolerable”. Journal of Pineal Research. 59 (4): 403–19. doi:10.1111/jpi.12267. PMID26272235. S2CID24373303. While originally thought to be produced exclusively in and secreted from the vertebrate pineal gland [53], it is now known that the indole is present in many, perhaps all, vertebrate organs [54] and in organs of all plants that have been investigated [48, 55, 56]. That melatonin is not relegated solely to the pineal gland is also emphasized by the reports that it is present in invertebrates [57–59], which lack a pineal gland and some of which consist of only a single cell.
^ Carrillo-Vico A, Guerrero JM, Lardone PJ, Reiter RJ (July 2005). “A review of the multiple actions of melatonin on the immune system”. Endocrine. 27 (2): 189–200. doi:10.1385/ENDO:27:2:189. PMID16217132. S2CID21133107.
^ Arushanian EB, Beĭer EV (2002). “[Immunotropic properties of pineal melatonin]”. Eksperimental’naia i Klinicheskaia Farmakologiia (in Russian). 65 (5): 73–80. PMID12596522.
^ Carrillo-Vico A, Reiter RJ, Lardone PJ, Herrera JL, Fernández-Montesinos R, Guerrero JM, Pozo D (May 2006). “The modulatory role of melatonin on immune responsiveness”. Current Opinion in Investigational Drugs. 7 (5): 423–31. PMID16729718.
^ Tan DX, Manchester LC, Liu X, Rosales-Corral SA, Acuna-Castroviejo D, Reiter RJ (March 2013). “Mitochondria and chloroplasts as the original sites of melatonin synthesis: a hypothesis related to melatonin’s primary function and evolution in eukaryotes”. Journal of Pineal Research. 54 (2): 127–38. doi:10.1111/jpi.12026. PMID23137057. S2CID206140413.
^ Sumi-Ichinose C, Ichinose H, Takahashi E, Hori T, Nagatsu T (March 1992). “Molecular cloning of genomic DNA and chromosomal assignment of the gene for human aromatic L-amino acid decarboxylase, the enzyme for catecholamine and serotonin biosynthesis”. Biochemistry. 31 (8): 2229–38. doi:10.1021/bi00123a004. PMID1540578.
^ Hickman AB, Klein DC, Dyda F (January 1999). “Melatonin biosynthesis: the structure of serotonin N-acetyltransferase at 2.5 A resolution suggests a catalytic mechanism”. Molecular Cell. 3 (1): 23–32. doi:10.1016/S1097-2765(00)80171-9. PMID10024876.
^ Lerner AB, Case JD, Takahashi Y (July 1960). “Isolation of melatonin and 5-methoxyindole-3-acetic acid from bovine pineal glands”. The Journal of Biological Chemistry. 235: 1992–7. PMID14415935.
^ Poeggeler B, Reiter RJ, Tan DX, Chen LD, Manchester LC (May 1993). “Melatonin, hydroxyl radical-mediated oxidative damage, and aging: a hypothesis”. Journal of Pineal Research. 14 (4): 151–68. doi:10.1111/j.1600-079X.1993.tb00498.x. PMID8102180. S2CID23460208.
^US patent 5449683, Wurtman RJ, “Methods of inducing sleep using melatonin”, issued 12 September 1995, assigned to Massachusetts Institute of Technology
^ Arendt J (August 2005). “Melatonin: characteristics, concerns, and prospects”. Journal of Biological Rhythms. 20 (4): 291–303. doi:10.1177/0748730405277492. PMID16077149. S2CID19011222. There is very little evidence in the short term for toxicity or undesirable effects in humans. The extensive promotion of the miraculous powers of melatonin in the recent past did a disservice to acceptance of its genuine benefits.
^ Reiter RJ (May 1991). “Pineal melatonin: cell biology of its synthesis and of its physiological interactions”. Endocrine Reviews. 12 (2): 151–80. doi:10.1210/edrv-12-2-151. PMID1649044. S2CID3219721.
^ Richardson GS (2005). “The human circadian system in normal and disordered sleep”. The Journal of Clinical Psychiatry. 66 Suppl 9: 3–9, quiz 42–3. PMID16336035.
^ Perreau-Lenz S, Pévet P, Buijs RM, Kalsbeek A (January 2004). “The biological clock: the bodyguard of temporal homeostasis”. Chronobiology International. 21 (1): 1–25. doi:10.1081/CBI-120027984. PMID15129821. S2CID42725506.
^ Jump up to:ab Arendt J, Skene DJ (February 2005). “Melatonin as a chronobiotic”. Sleep Medicine Reviews. 9 (1): 25–39. doi:10.1016/j.smrv.2004.05.002. PMID15649736. Exogenous melatonin has acute sleepiness-inducing and temperature-lowering effects during ‘biological daytime’, and when suitably timed (it is most effective around dusk and dawn), it will shift the phase of the human circadian clock (sleep, endogenous melatonin, core body temperature, cortisol) to earlier (advance phase shift) or later (delay phase shift) times.
^ Chaturvedi CM (1984). “Effect of Melatonin on the Adrenl and Gonad of the Common Mynah Acridtheres tristis”. Australian Journal of Zoology. 32 (6): 803–09. doi:10.1071/ZO9840803.
^ Chen HJ (July 1981). “Spontaneous and melatonin-induced testicular regression in male golden hamsters: augmented sensitivity of the old male to melatonin inhibition”. Neuroendocrinology. 33 (1): 43–6. doi:10.1159/000123198. PMID7254478.
^ Paredes SD, Korkmaz A, Manchester LC, Tan DX, Reiter RJ (1 January 2009). “Phytomelatonin: a review”. Journal of Experimental Botany. 60 (1): 57–69. doi:10.1093/jxb/ern284. PMID19033551. S2CID15738948.
^ Bonnefont-Rousselot D, Collin F (November 2010). “Melatonin: action as antioxidant and potential applications in human disease and aging”. Toxicology. 278 (1): 55–67. doi:10.1016/j.tox.2010.04.008. PMID20417677.
^ Reiter RJ, Manchester LC, Tan DX (September 2005). “Melatonin in walnuts: influence on levels of melatonin and total antioxidant capacity of blood”. Nutrition. 21 (9): 920–4. doi:10.1016/j.nut.2005.02.005. PMID15979282.
^ Burkhardt S, Tan DX, Manchester LC, Hardeland R, Reiter RJ (October 2001). “Detection and quantification of the antioxidant melatonin in Montmorency and Balaton tart cherries (Prunus cerasus)”. Journal of Agricultural and Food Chemistry. 49 (10): 4898–902. doi:10.1021/jf010321. PMID11600041.
^ Lamont KT, Somers S, Lacerda L, Opie LH, Lecour S (May 2011). “Is red wine a SAFE sip away from cardioprotection? Mechanisms involved in resveratrol- and melatonin-induced cardioprotection”. Journal of Pineal Research. 50 (4): 374–80. doi:10.1111/j.1600-079X.2010.00853.x. PMID21342247. S2CID8034935.
^ Hattori A, Migitaka H, Iigo M, Itoh M, Yamamoto K, Ohtani-Kaneko R, et al. (March 1995). “Identification of melatonin in plants and its effects on plasma melatonin levels and binding to melatonin receptors in vertebrates”. Biochemistry and Molecular Biology International. 35(3): 627–34. PMID7773197.
^ Sae-Teaw M, Johns J, Johns NP, Subongkot S (August 2013). “Serum melatonin levels and antioxidant capacities after consumption of pineapple, orange, or banana by healthy male volunteers”. Journal of Pineal Research. 55 (1): 58–64. doi:10.1111/jpi.12025. PMID23137025. S2CID979886.
^ Rodriguez RR (13 January 2010). “Warning Letter”. Inspections, Compliance, Enforcement, and Criminal Investigations. U.S. Food and Drug Administration. Archived from the original on 12 January 2017.
^Bebida Beverage Company U.S. Food and Drug Administration 4 March 2015 (Accessed 8 December 2017)
^ Miroddi M, Bruno R, Galletti F, Calapai F, Navarra M, Gangemi S, Calapai G (March 2015). “Clinical pharmacology of melatonin in the treatment of tinnitus: a review”. European Journal of Clinical Pharmacology. 71 (3): 263–70. doi:10.1007/s00228-015-1805-3. PMID25597877. S2CID16466238.
^ Peres MF, Masruha MR, Zukerman E, Moreira-Filho CA, Cavalheiro EA (April 2006). “Potential therapeutic use of melatonin in migraine and other headache disorders”. Expert Opinion on Investigational Drugs. 15 (4): 367–75. doi:10.1517/13543784.15.4.367. PMID16548786. S2CID28114683.
^ Mills E, Wu P, Seely D, Guyatt G (November 2005). “Melatonin in the treatment of cancer: a systematic review of randomized controlled trials and meta-analysis”. Journal of Pineal Research. 39 (4): 360–6. doi:10.1111/j.1600-079X.2005.00258.x. PMID16207291. S2CID22225091.
^ Meltz ML, Reiter RJ, Herman TS, Kumar KS (March 1999). “Melatonin and protection from whole-body irradiation: survival studies in mice”. Mutation Research. 425 (1): 21–7. doi:10.1016/S0027-5107(98)00246-2. PMID10082913.
^ Reiter RJ, Herman TS, Meltz ML (December 1996). “Melatonin and radioprotection from genetic damage: in vivo/in vitro studies with human volunteers”. Mutation Research. 371 (3–4): 221–8. doi:10.1016/S0165-1218(96)90110-X. PMID9008723.
^ Reiter RJ, Herman TS, Meltz ML (February 1998). “Melatonin reduces gamma radiation-induced primary DNA damage in human blood lymphocytes”. Mutation Research. 397 (2): 203–8. doi:10.1016/S0027-5107(97)00211-X. PMID9541644.
^ Tan DX, Manchester LC, Terron MP, Flores LJ, Reiter RJ (January 2007). “One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species?”. Journal of Pineal Research. 42 (1): 28–42. doi:10.1111/j.1600-079X.2006.00407.x. PMID17198536. S2CID40005308.
^ Giannotti F, Cortesi F, Cerquiglini A, Bernabei P (August 2006). “An open-label study of controlled-release melatonin in treatment of sleep disorders in children with autism”. Journal of Autism and Developmental Disorders. 36 (6): 741–52. doi:10.1007/s10803-006-0116-z. PMID16897403. S2CID19724241.
^ Bendz LM, Scates AC (January 2010). “Melatonin treatment for insomnia in pediatric patients with attention-deficit/hyperactivity disorder”. The Annals of Pharmacotherapy. 44(1): 185–91. doi:10.1345/aph.1M365. PMID20028959. S2CID207263711.
CitrullineCAS Registry Number: 372-75-8 CAS Name:N5-(Aminocarbonyl)-L-ornithine Additional Names: d-ureidonorvaline; a-amino-d-ureidovaleric acid; Nd-carbamylornithine Molecular Formula: C6H13N3O3Molecular Weight: 175.19 Percent Composition: C 41.13%, H 7.48%, N 23.99%, O 27.40%Line Formula: H2NCONH(CH2)3CH(NH2)COOH Literature References: An amino acid, first isolated from the juice of watermelon, Citrullus vulgaris Schrad., Cucurbitaceae: Wada, Biochem. Z.224, 420 (1930); isoln from casein: Wada, ibid.257, 1 (1933). Synthesis from ornithine through copper complexes: Kurtz, J. Biol. Chem.122, 477 (1938); by alkaline hydrolysis of arginine: Fox, ibid.123, 687 (1938); from cyclopentanone oxime: Fox et al.,J. Org. Chem.6, 410 (1941). Crystallization: Matsuda et al.,JP71 174 (1971 to Ajinomoto), C.A.74, 126056u (1971). Crystal and molecular structure: Naganathan, Venkatesan, Acta Crystallogr.27B, 1079 (1971); Ashida et al.,ibid.28B, 1367 (1972). Use in asthenia and hepatic insufficiency: FR2198739 (1974 to Hublot & Vallet), C.A.82, 144952c (1975). Clinical trial in treatment of lysinuric protein intolerance: J. Rajantie et al.,J. Pediatr.97, 927 (1980); T. O. Carpenter et al.,N. Engl. J. Med.312, 290 (1985).Properties: Prisms from methanol + water, mp 222°. [a]D20 +3.7° (c = 2). pK1 2.43; pK2 9.41. Sol in water. Insol in methanol, ethanol.Melting point: mp 222°pKa: pK1 2.43; pK2 9.41Optical Rotation: [a]D20 +3.7° (c = 2) Derivative Type: HydrochlorideCAS Registry Number: 34312-10-2Molecular Formula: C6H13N3O3.HClMolecular Weight: 211.65Percent Composition: C 34.05%, H 6.67%, N 19.85%, O 22.68%, Cl 16.75%Properties: Crystals, dec 185°. [a]D22 +17.9° (c = 2).Optical Rotation: [a]D22 +17.9° (c = 2) Derivative Type: Malate (salt)CAS Registry Number: 54940-97-5Trademarks: Stimol (Biocodex)Molecular Formula: C6H13N3O3.C4H6O5Molecular Weight: 309.27Percent Composition: C 38.84%, H 6.19%, N 13.59%, O 41.39% Therap-Cat: Treatment of asthenia.
Asklepion is developing an iv formulation of citrulline, Citrupress, for the potential treatment of pulmonary hypertension and for the potential prevention of clinical sequelae of acute lung injury complicating congenital heart repair surgery in pediatric patients, and also investigating the drug for the potential treatment of acute sickle cell crisis. In August 2016, a phase III study was initiated for preventing clinical sequelae of acute lung injury?in pediatric patients undergoing cardiopulmonary bypass (CPB) for heart defects; in July 2019, results were expected in October 2019.
Citrulline is an amino acid. It is made from ornithine and carbamoyl phosphate in one of the central reactions in the urea cycle. It is also produced from arginine as a by-product of the reaction catalyzed by NOS family. Its name is derived from citrullus, the Latin word for watermelon, from which it was first isolated.
Citrulline is made from ornithine and carbamoyl phosphate in one of the central reactions in the urea cycle. It is also produced from arginine as a byproduct of the reaction catalyzed by NOS family (NOS; EC 1.14.13.39).[6] It is made from arginine by the enzymetrichohyalin at the inner root sheath and medulla of hair follicles.[7]Arginine is first oxidized into N-hydroxyl-arginine, which is then further oxidized to citrulline concomitant with release of nitric oxide.
Citrulline is also made by enterocytes of the small intestine.[2][8]
Function
Several proteins contain citrulline as a result of a posttranslational modification. These citrulline residues are generated by a family of enzymes called peptidylarginine deiminases (PADs), which convert arginine into citrulline in a process called citrullination or deimination with the help of calcium ion. Proteins that normally contain citrulline residues include myelin basic protein (MBP), filaggrin, and several histone proteins, whereas other proteins, such as fibrin and vimentin are susceptible to citrullination during cell death and tissue inflammation.
Circulating citrulline concentration is a biomarker of intestinal functionality.[9][10
PAPER
Biochemistry, 53(41), 6511-6519; 2014
PAPER
Journal of the Chemical Society of Pakistan, 34(2), 451-454; 2012
PAPER
Journal of Agricultural and Food Chemistry, 66(33), 8841-8850; 2018
l-Citrulline is a nonessential amino acid with a variety of physiological functions and can be enzymatically produced by arginine deiminase (ADI, EC 3.5.3.6). The enzymatic-production approach is of immense interest because of its mild conditions, high yield, low cost, and environmental benignity. However, the major hindrances of l-citrulline industrialization are the poor thermostability and enzyme activity of ADI. Hence, in this work, directed evolution and site-directed mutagenesis aided with in silico screening, including the use of b-factor values and HoTMuSiC, were applied to a previously identified ADI from Enterococcus faecalis SK23.001 (EfADI), and a triple-site variant R15K–F269Y–G292P was obtained. The triple-site variant displays a 2.5-fold higher specific enzyme activity (333 U mg–1), a lower Km value of 6.4 mM, and a 6.1-fold longer half-life (t1/2,45°C = 86.7 min) than wild-type EfADI. This work provides a protein-engineering strategy to improve enzyme activity and thermostability, which might be transferrable to other ADIs and enzymes.
Biocatalytic transformation of carbamate formed readily from CO2 and NH3 provides attractive green routes for mitigation of these important environmental pollutants. Accordingly, a coupled-enzyme system was developed for the one-pot production of citrulline through carbamoylation of ornithine in aqueous solutions of CO2 and NH3. Hyperthermophilic ornithine carbamoyltransferases are produced recombinantly in E. coli with carbamate kinases known to have a propensity for carbamoyl phosphate synthesis. Importantly, in vitro biocatalysis is carried out by E. coli cell lysate prepared through coexpression of the required recombinant enzymes in a single bacterial culture, greatly reducing limitations normally associated with protein production and purification. Acetate kinase that is endogenous in the lysate also recycles the required ATP cofactor, which would otherwise have been required in costly stoichiometric amounts. Recombinant lysates catalyze the production of carbamoyl phosphate with substoichiometric ATP (>300 turnovers) as well as its in situ reaction with ornithine to give citrulline in high yield (>95%) and g L–1 h–1 titers. The system is active over a wide range of NH3 concentrations (2.5 mM – 2 M), and >90% conversions of NH3 may be reached within 1.5 h. Aqueous NH3 used to sequester CO2 gas (10% v/v) may be directly used as the biocatalyst feedstock. In preliminary studies, citrulline is found to be an effective organic nitrogen fertilizer of the wheat grass Brachypodium distachyon. Therefore, lysates described here constitute a cost-effective biocatalytic platform for one-pot production of a promising organic nitrogen fertilizer, under mild reaction conditions, from environmental pollutants as feedstock.
Process for preparing citrulline from a transition metal complex of ornithine using cyanate useful to reduce the incidence or severity of cardiopulmonary bypass-induced pulmonary injury due to free radical formation in a patient during cardiopulmonary bypass.
Ornithine is an alpha amino acid with a terminal amino group opposite the alpha carbon.
Citrulline is an alpha amino acid with a terminal carbamido group in the same position as the terminal amino group of ornithine. Dr. A. Kurtz described synthesis of racemic citrulline from racemic ornithine in 1938 (J. Biol. Chem., 122:477-484), and that disclosure was followed up by synthesis of optically active /-citrulline from /-ornithine in 1949 (J. Biol. Chem., 180: 1253-1267). Optical activity was preserved by complexing the starting material (/-ornithine) in a transition metal complex via the alpha amino and carboxyl groups, then reacting the terminal amino group with urea to from a carbamido derivative (see Figure 1). Kurth 1949 describes numerous other syntheses, all depending on the transition metal complex to preserve the alpha amino acid character of the starting compound while derivatizing other parts of the molecule. An example of this synthesis is described in Example 1 below.
Details of various steps in the improved processes developed by the present inventors for producing pharmaceutical grade citrulline are discussed below.
Synthesis of Citrulline from Ornithine
[00014] The present inventors preserved the stereochemical structure around the alpha carbon of the alpha amino acid during reaction of amino groups elsewhere on the compound by complexing the alpha end of the molecule with a transition metal atom, as reported by
Kurth 1938 and 1949. The initial production of the /-ornithine-copper complex is carried out as described by Kurtz. Kurtz describes a variety of transition metals as the complexing metal in the 1949 paper, but the preferred metal is copper (II), based on the ease of forming stable complexes and the ease with which copper (II) may subsequently be removed from the product. The copper is typically supplied as cupric sulfate, although complex formation from copper (II) acetate, cupric carbonate, or cupric oxide have also been reported.
[00015] The present inventors have discovered an alternative method of derivatizing the terminal amino group of the complexed alpha amino acid using cyanate rather than the urea reaction reported by Kurth. An example of this improved synthesis is shown in Figure 2A and described in Example 3 below. Use of cyanate as the derivatizing agent has been found to produce fewer distinct product compounds, which simplifies purification of the desired citrulline product. Kurth carried out urea derivatization by refluxing the copper complex in the presence of excess urea. Cyanate derivatization may be carried out at lower temperatures (e.g. 55°C-65°C) which may contribute to higher yield of citrulline, based on the initial amount of ornithine. Cyanate is preferably provided in excess, and the reaction is driven by precipitation of the citrulline: copper complex. The precipitated complex is washed with water to remove unreacted copper (e.g., wash until no blue coloration persists in the filtrate). The precipitated copper complex of citrulline may be recovered and dried.
Enriching Citrulline as a Copper Complex
[00016] The inventors have discovered that the relative citrulline content of the reaction
product(s) can be enhanced by reprecipitation of the citrulline: copper complex. Precipitated copper complex of citrulline (produced, for example, by reaction of a ornithine: copper complex with urea or cyanate in water) may be dried. The
citrulline: copper complex may be redissolved by suspending the precipitate in water and acidifying the suspension until the complex dissolves. Acidification may be
accomplished by adding concentrated acid, preferably hydrogen chloride, to the suspension while stirring. Once the copper: citrulline complex solution is clear, base (typically sodium hydroxide) is added to bring the pH up to 7-10. Both the acidification and subsequent neutralization steps are actively cooled (temperature not more than 45°C) to protect the citrulline product from hydrolysis or reaction to produce side products. The precipitate is washed with water (e.g., until the filtrate is free of chloride by checking the filtrate for turbidity with silver nitrate), and then the precipitate is dried. Reprecipitation under these conditions is selective for citrulline: copper complex over ornithine: copper complex, because the ornithine complex is more soluble in water. If the dried complex contains higher than the desired level of ornithine contamination (e.g., greater than 10 mole% ornithine – as measured by NMR, for example), the complex may be redissolved and reprecipitated as necessary to further lower the relative amount of ornithine.
Recovering Citrulline from Its Copper Complex
[00017] Once the ornithine content in the copper: citrulline complex precipitate is sufficiently low
(preferably less than 10 mole% ornithine), the precipitate is resuspended in water and citrulline is freed from the complex by removing the copper as an inorganic precipitate, typically copper sulfide (See Figure 2B). Sulfide may be introduced in a variety of salt forms, but the inventors have found it preferable to use hydrogen sulfide gas as the sulfide source. In a preferred mode, the aqueous suspension is placed in a stirred, pressure vessel. The air is then pumped out of the reactor’s head space to form an under pressure. The reactor is then repressurized with hydrogen sulfide gas over the aqueous suspension (preferably at low temperature, e.g., 0°C-5°C, to maximize the solubility of hydrogen sulfide). Hydrogen sulfide is continuously added to the reactor to maintain parity with ambient pressure during consumption of this gas. Copper salts will precipitate, leaving citrulline in solution. As hydrogen sulfide is consumed, the pressure in the vessel decreases; the reaction is complete when the pressure stabilizes. Reaction of hydrogen sulfide with residual copper salts (for example chloride or sulfate) will lower the pH; typically the pH will be below 4, preferably pH~3. Copper salts typically include copper (II) sulfide, but may also include copper (I) sulfide and copper oxide. The solution temperature is elevated for filtration, typically to about 30°C, to promote solubility of the citrulline and drive off excess hydrogen sulfide gas, while precipitated copper salts are removed by filtration.
Purifying Citrulline
[00018] For pharmaceutical use, the active compound must be substantially free of contaminants, and further purification steps are necessary to produce a pharmaceutical grade product. For the purposes of this invention, substantially free of contaminants is considered to include: ornithine not more than (NMT) 0.8%, individual specified impurities NMT 0.15%, individual unspecified (unknown) impurities NMT 0.1%; total related substances NMT 1.3%, and Cu not more than lOppm. For citrulline manufactured from ornithine using copper complex to protect the alpha amino acid functions, the inventors have found that desired purification after citrulline is released from the copper complex can be achieved by activated carbon adsorption of contaminants and solvent/anti- solvent crystallization of the active pharmaceutical component.
[00019] The citrulline-containing aqueous solution remaining after removal of precipitated copper salts is neutralized to stabilize the citrulline against hydrolysis, to enhance adsorption of residual copper to activated carbon, and to facilitate solvent/anti-solvent precipitation of citrulline; pH is preferably adjusted to 5.9 ± 0.2, the isoelectric point of citrulline. The neutralized citrulline solution may be passed through a nano-filter to remove any bacteria and/or bacterial cell wall fragments that contaminate the solution. The nano-filtered solution may be held in a semi-sterile reservoir for staging purposes between the subsequent purification steps. The neutralized citrulline solution is treated with activated carbon, either by mixing with carbon dust or passing the solution through an activated carbon adsorber bed. The aqueous citrulline-containing effluent from the activated carbon is mixed with an anti-solvent to induce anti-solvent crystallization. Suitable anti solvents are miscible with water, including aliphatic alcohols, such as 2-propanol, ethanol or methanol, as well as acetone. A preferred antisolvent for citrulline is acetone, when mixed with approximately two volumes of water (e.g., 1 volume of water to 1.8 volumes of acetone). Acetone is preferably pre-cooled so that the resultant suspension is 0°C- 10°C. The cooled suspension may be collected in a reservoir or processed by filtration immediately to recover the citrulline precipitate.
Microbial control:
[00020] Because citrulline synthesis and purification occur in aqueous solution, there is increased risk of microbial contamination and endotoxin accumulation in the product. Washing the citrulline: copper precipitate, and addition of H2S to acid solution minimize any accumulation of microbes. From the exposure of the complex to FES until treatment with acetone the aqueous solutions of citrulline are preferably kept in sealed vessels to limit microbial contamination and growth. Enclosing the purification steps to minimize contact with the environment and use of sterile filters to capture potential microbial contamination allows the manufacturing to be performed in an ISO 8 cleanroom. Alternatively, the final purification steps can be carried out in a sterile GMP environment of the sort used for aseptic filling of sterile dosage products (e.g., ISO Class 5/6).
[00021] If examination of the solution prior to the anti-solvent precipitation shows the amounts of microbes or endotoxin levels exceed those aceptable for injectable therapeutic compositions (e.g., 50 EU/g API, more preferably 20 EU/g), the product may be subjected to nano-filtration to remove microbes and endotoxin, before being recovered by anti-solvent precipitation and drying. The citrulline and water molecules pass through the nano-filtration membrane, but the larger bacteria and bacterial cell wall fragments are retained by the filter.
Filter press
[00022] The reaction mixtures may be pumped through a filter press to collect / remove the
suspended solids. See the general picture in Figure 3, and the attached photograph in Figure 4. The press is composed of a series of plates 1 which are then hydraulically pressed together. The hydraulic pressure ensures that the system is sealed. The suspension is then pumped through a central tube 2 where it spreads-out across several chambers 3 between the plates. The walls of the plates have a filter sheet, which allows the filtrate to flow past and exit via an internal cavity 4.
[00023] The general advantage of a filter press is that it allows a high surface area for filtration.
This effect greatly accelerates the portion-wise collection and washing of the complex and API. This system may be used to collect the copper salts after exposure to hydrogen sulfide. In the latter case, the suspension is pumped from the reactor into the press, and the filtrate may then be passed through an in-line 5 pm filter to catch any residual particulate copper, then an in-line sterile 0.2 pm filter at the entry port of a semi-sterile container for holding.
The press may be used to collect:
• Crude citrulline copper complex
• The complex after the pH-driven re-precipitation
• Precipitated copper salts (where citrulline leaves as solution in the filtrate)
• Precipitated citrulline from anti-solvent precipitation prior to drying
Semi-sterile containers
[00024] A useful semi-sterile container is basically a closed vessel equipped with a stirrer and ports for the addition and removal of liquid, and a pH meter. The container should be sterilized (e.g., treated with isopropyl alcohol solution and rinsed with water) directly prior to use and not opened during use. A sterile, air filter attached to the lid allows air to flow into the container as the liquid is being pumped out. The pH adjustment may be performed in this container, before treatment with activated carbon. The container is not particularly suitable for the long-term storage of the solutions.
Activated carbon adsorber bed
[00025] The solution may be pumped from the semi sterile container through the activated carbon bed (a column packed with granulated activated carbon) pre-flushed with argon. The liquid is then returned to the semi-sterile container via an in-line 5 pm filter and the 0.2 pm sterile filter at the entry port. If the solution is pumped in a cyclic manner with the stirrer activated for not less than 6 hours, the sterile filter acts as a“microbial scrubber” continually collecting any microbes in the solution. The activated carbon primarily removes any organic impurities and will also remove any residual dissolved copper ions. The 5 pm filter catches any carbon particles which detach from the bed.
Sterile bags
[00026] After processing in the activated carbon adsorber bed, the solution may be passed into a single use sterile bag via another sterile filter. The solution may be stored longer in the bag than in the semi-sterile container. At this point, a test for the presence of microbes and/or bacterial endotoxins can be carried out. If endotoxins are observed, then the cut off (nano-filtration) membrane may be employed. If not, the citrulline is ready to be
recovered from the solution by anti-solvent precipitation. Collection of the solution in a sterile bag allows the citrulline solution to be processed batch-wise, where conveniently sized portions of citrulline are precipitated and recovered in the filter press.
Solvent/Anti-solvent Mixing
[00027] The aqueous citrulline solution is mixed with pre-cooled anti-solvent to precipitate the citrulline from solution. After mixing with anti-solvent, the threat posed by bacterial growth is not higher than that for other APIs. The addition of the organic solvent makes the resulting solution bacteriostatic at a minimum. This precipitation improves the purity of citrulline, reducing, in particular, the ornithine levels, and allows for the rapid extraction of citrulline from solution.
Final drying
[00028] The precipitate is dried to remove residual acetone and water. Drying may be carried-out in a conical dryer, firstly to drive off the acetone anti-solvent, then moisture and finally the water of crystallization. The conical dryer can also be used to homogenize the product. The final, dry product of anti-solvent precipitation may be stored, and ultimately dissolved in sterile aqueous diluent for therapeutic administration.
[00029] On dissolution in sterile aqueous media, citrulline prepared as described herein may be used to treat pulmonary hypertension (WO/2000/073322), bronchopulmonary dysplasia (WO/2009/099998), sickle cell crisis (WO/2018/157137), cardiac surgery patients (WO/2005/082042), cardiopulmonary bypass patients (WO/2018/125999), and vasospasm as a complication of subarachnoid hemorrhage (WO/2009/099999), by parenteral administration as described in these documents, incorporated herein by reference.
EXAMPLES
Example 1. Synthesis of citrulline from ornithine using urea.
[00030] L-Citrulline is synthesized from L-omithine and urea. A flow chart of the reaction is shown in Figure 1 A.
[00031] L-Citrulline is prepared synthetically starting from L-ornithine hydrochloride. Into a 120- L reactor containing approximately 50 liters of water, 10 kilograms of L-omithine hydrochloride is added and dissolved. The solution is neutralized with potassium hydroxide and then converted to its copper complex by the addition of 15kg copper sulfate (molar equivalent amount). The copper complex protects the 2-amino carboxylic acid functionality in the molecule while chemistry is performed on the terminal amino group. The L-ornithine copper complex is then exposed to an excess of urea at reflux, which promotes its conversion to the copper complex of L-citrulline. The resulting copper complex of L-citrulline then is precipitated and collected by filtration.
[00032] The isolated copper complex of L-citrulline is dried and testing is performed. The
appearance is verified, and an in-use performance test is done to determine suitability to proceed.
Example 2. Purification of citrulline from copper-citrulline complex.
[00033] L-Citrulline synthesized from L-ornithine and urea is purified by resin-based purification and recrystallization. A flow chart of the reaction is shown in Figure IB.
[00034] In a 120-L reactor, ~13 kilograms of the L-citrulline copper complex prepared in
Example 1 is added to a stirring solution of sodium sulfide (Na2S) in water
(approximately 8 kilograms Na2S in 50 liters of water), causing the precipitation of copper sulfide and the freeing of L-citrulline. The solution is filtered to remove the copper salts. The pH of the resulting aqueous solution containing the sodium salt of L- citrulline and residual sodium sulfide is lowered to 4 by the addition of an acidic ion exchange resin (such as Amberlite). A constant stream of argon gas is passed through the solution to remove the residual sulfide as hydrogen disulfide. The pH of the solution is then raised to 5.9 ± 0.2 using sodium hydroxide to form isoelectric L-citrulline.
Activated carbon is then added to the reaction mixture to remove residual impurities, in particular residual copper ions. The solids (Amberlite and activated carbon) are then removed by filtration, and the filtrate is concentrated to approximately 50 liters (either by evaporation or reverse osmosis). L-citrulline is then precipitated from the aqueous solution by the addition of an equal part of acetone, and the mixture is cooled to near 0°C. The precipitate is collected by filtration and dried in a vacuum oven.
[00035] The non-sterile bulk powder is then reconstituted and processed for endotoxin reduction and sterile filtration steps followed by crystallization, drying and micronization in an aseptic environment. The sterile bulk powder is then used as the“raw material” for aseptic filling into glass vials to produce the finished drug product which may be reconstituted with a sterile diluent prior to use.
Example 3. Synthesis of citrulline from ornithine using cyanate
[00036] L-Citrulline was prepared synthetically starting from L-omithine hydrochloride. Into a reactor containing sodium hydroxide (11 kg) in water (170 kg), L-ornithine hydrochloride (44 kg) was added and dissolved. The temperature was maintained at no more than 40°C by active cooling. The ornithine was then converted to its copper complex by the addition of 0.5 molar equivalents of copper sulfate (33 kg) and stirring at ambient temperature for more than 15 minutes. The copper complex protects the 2-amino carboxylic acid functionality of the molecule while chemistry is performed on the terminal amino group. A molar excess of potassium cyanate (32 kg) is then added to the L-ornithine copper complex, and the solution is held at 55°C-65°C for 4.0-4.5 hours, which promotes its conversion to the copper complex of L-citrulline. The resulting copper complex of L-citrulline precipitates during the reaction, and it is collected by filtration.
Example 4. Purification of therapeutic grade citrulline.
[00037] The dry copper: citrulline complex produced in Example 3 is added to a reactor
containing water, which is stirred to resuspend the complex. Concentrated hydrogen chloride solution is added to convert the complex into a solution of copper (II) chloride and citrulline hydrochloride, while the temperature of the reactor is maintained at no more than 45°C by active cooling. Once the contents of the reactor are in solution, sodium hydroxide is added to raise the pH to 7-10, while the temperature is maintained at no more than 40°C. The copper complex of citrulline then precipitates. The precipitate is collected and washed with water until no blue coloration persists in the filtrate.
[00038] The washed precipitate is tested to determine the relative ornithine content. If ornithine is greater than 10 mole%, the precipitate is redissolved and resuspended as described above, until the ornithine content is lowered to not more than 10 mole%.
[00039] Once the precipitate achieves the desired ornithine content, it is resuspended in water in a stirred reactor, and hydrogen sulfide gas is introduced into the suspension to precipitate copper sulfide and dissolve citrulline. The solution is warmed to 30°C ± 2°C to ensure citrulline is fully solubilized, and precipitated copper salts are removed by filtration. The citrulline-containing filtrate is passed thorough micro- and sterile-filtrations and collected in a semi-sterile reactor.
[00040] Activated carbon is used to remove residual impurities, in particular an organic
component and residual copper ions. The pH of the resulting aqueous solution containing L-citrulline and residual copper is adjusted to 5.9 ± 0.2 with sodium hydroxide to form isoelectric citrulline solution. The isoelectric citrulline solution is treated with active carbon granules, preferably by passing the solution through an active carbon adsorber bed, and passed through micro and sterile filters after the active carbon treatment.
[00041] L-citrulline is then precipitated from the aqueous solution by the addition of acetone anti solvent, and the mixture is cooled to near 0°C. Addition of 1.5 to 2 volume equivalents of acetone produce dihydrate crystals of citrulline. The precipitate is collected by filtration. The crystals are dried in a vacuum in a conical dryer at temperature of no more than 45°C to remove acetone and water, resulting in an anhydrous crystalline solid. This solid citrulline corresponds to the orthorhombic d form anhydrous crystals reported by Allouchi, et al., 2014 ( Cryst . Growth Des., 14: 1279-1286).
[00042] Either the dihydrate crystals or the anhydrous crystals may be used therapeutically. The solid or an aqueous solution/suspension may be administered enterally, or the solid may be redissolved for parenteral administration. To produce a final therapeutic product, the non-sterile bulk powder was reconstituted and underwent endotoxin reduction and sterile filtration steps followed by crystallization, drying and micronization in an aseptic environment. The sterile bulk powder was then used as the“raw material” for aseptic filling into glass vials to produce the finished drug product which was reconstituted with a sterile diluent prior to use.
References
^“Citrulline – Compound Summary”. PubChem Compound. USA: National Center for Biotechnology Information. 16 September 2004. Identification. Retrieved 1 May 2012.
^ Rogers, G. E.; Rothnagel, J. A. (1983). “A sensitive assay for the enzyme activity in hair follicles and epidermis that catalyses the peptidyl-arginine-citrulline post-translational modification”. Current Problems in Dermatology. 11: 171–184. doi:10.1159/000408673. ISBN978-3-8055-3752-0. PMID6653155.
^ Crenn, P.; et al. (2000). “Post-absorptive plasma citrulline concentration is a marker of intestinal failure in short bowel syndrome patients”. Gastroenterology. 119 (6): 1496–505. doi:10.1053/gast.2000.20227. PMID11
The U.S. Food and Drug Administration approved Ebanga (Ansuvimab-zykl), a human monoclonal antibody, for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga blocks binding of the virus to the cell receptor, preventing its entry into the cell.
Zaire ebolavirus is one of four Ebolavirus species that can cause a potentially fatal human disease. It is transmitted through blood, body fluids, and tissues of infected people or wild animals, and through surfaces and materials, such as bedding and clothing, contaminated with these fluids. Individuals who care for people with the disease, including health care workers who do not use correct infection control precautions, are at the highest risk for infection.
During an Ebola outbreak in the Democratic Republic of the Congo (DRC) in 2018-2019, Ebanga was evaluated in a clinical trial (the PALM trial). The PALM trial was led by the U.S. National Institutes of Health and the DRC’s Institut National de Recherche Biomédicale with contributions from several other international organizations and agencies.
In the PALM trial, the safety and efficacy of Ebanga was evaluated in a multi-center, open-label, randomized controlled trial. 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. The primary analysis population was all patients who were randomized and concurrently eligible to receive either Ebanga or the investigational control during the same time period of the trial. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
The most common symptoms experienced while receiving Ebanga include: fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection. Hypersensitivity, including infusion-related events, can occur in patients taking Ebanga, and treatment should be discontinued in the event of a hypersensitivity reaction.
Patients who receive Ebanga should avoid the concurrent administration of a live virus vaccine against Ebolavirus. There is the potential for Ebanga to inhibit replication of a live vaccine virus and possibly reduce the efficacy of this vaccine.
FDA granted the approval to Ridgeback Biotherapeutics, LP.
Ansuvimab, sold under the brand name Ebanga, is a monoclonal antibody medication for the treatment of Zaire ebolavirus (Ebolavirus) infection.[1][2]
The most common symptoms include fever, tachycardia (fast heart rate), diarrhea, vomiting, hypotension (low blood pressure), tachypnea (fast breathing) and chills; however, these are also common symptoms of Ebolavirus infection.[1]
Ansuvimab was approved for medical use in the United States in December 2020.[1][2]
Ansuvimab is a monoclonal antibody therapy that is infused intravenously into patients with Ebola virus disease. Ansuvimab is a neutralizing antibody,[3] meaning it binds to a protein on the surface of Ebola virus that is required to infect cells. Specifically, ansuvimab neutralizes infection by binding to a region of the Ebola virus envelope glycoprotein that, in the absence of ansuvimab, would interact with virus’s cell receptor protein, Niemann-Pick C1 (NPC1).[6][7][8] This “competition” by ansuvimab prevents Ebola virus from binding to NPC1 and “neutralizes” the virus’s ability to infect the targeted cell.[6]
Effector function
Antibodies have antigen-binding fragment (Fab) regions and constant fragment (Fc) regions. The Neutralization of virus infection occurs when the Fab regions of antibodies binds to virus antigen(s) in a manner that blocks infection. Antibodies are also able to “kill” virus particles directly and/or kill infected cells using antibody-mediated “effector functions” such as opsonization, complement-dependent cytotoxicity, antibody-dependent cell-mediated cytotoxicity and antibody-dependent phagocytosis. These effector functions are contained in the Fc region of antibodies, but is also dependent on binding of the Fab region to antigen. Effector functions also require the use of complement proteins in serum or Fc-receptor on cell membranes. Ansuvimab has been found to be capable of killing cells by antibody-dependent cell-mediated cytotoxicity.[3] Other functional killing tests have not been performed.
Ansuvimab has also shown success with lowering the mortality rate from ~70% to about 34%. In August 2019, Congolese health authorities, the World Health Organization, and the U.S. National Institutes of Health promoted the use of ansuvimab, alongside REGN-EB3, a similar Regeneron-produced monoclonal antibody treatment, over other treatments yielding higher mortality rates, after ending clinical trials during the outbreak.[13][14]
In an experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and treated with a combination of ansuvimab and another antibody isolated from the same subject, mAb100. Three doses of the combination were given once a day starting 1 day after the animals were infected. The control animal died and the treated animals all survived.[3]
Ansuvimab monotherapy
In a second experiment described in the 2016 paper, rhesus macaques were infected with Ebola virus and only treated with ansuvimab. Three doses of ansuvimab were given once a day starting 1 day or 5 days after the animals were infected. The control animals died and the treated animals all survived.[3] Unpublished data referred to in a publication of the 2018 Phase I clinical trial results of ansuvimab, reported that a single infusion of ansuvimab provided full protection of rhesus macaques and was the basis of the dosing used for human studies.[5][4]
Experimental use in the Democratic Republic of Congo
During the 2018 Équateur province Ebola outbreak, ansuvimab was requested by the Democratic Republic of Congo (DRC) Ministry of Public Health. Ansuvimab was approved for compassionate use by the World Health OrganizationMEURI ethical protocol and at DRC ethics board. Ansuvimab was sent along with other therapeutic agents to the outbreak sites.[19][20][11] However, the outbreak came to a conclusion before any therapeutic agents were given to patients.[11]
Approximately one month following the conclusion of the Équateur province outbreak, a distinct outbreak was noted in Kivu in the DRC (2018–20 Kivu Ebola outbreak). Once again, ansuvimab received approval for compassionate use by WHO MEURI and DRC ethic boards and has been given to many patients under these protocols.[11] In November 2018, the Pamoja Tulinde Maisha (PALM [together save lives]) open-label randomized clinical control trial was begun at multiple treatment units testing ansuvimab, REGN-EB3 and remdesivir to ZMapp. Despite the difficulty of running a clinical trial in a conflict zone, investigators have enrolled 681 patients towards their goal of 725. An interim analysis by the Data Safety and Monitoring Board (DSMB) of the first 499 patient found that ansuvimab and REGN-EB3 were superior to the comparator ZMapp. Overall mortality of patients in the ZMapp and remdesivir groups were 49% and 53% compared to 34% and 29% for ansuvimab and REGN-EB3. When looking at patients who arrived early after disease symptoms appeared, survival was 89% for ansuvimab and 94% for REGN-EB3. While the study was not powered to determine whether there is any difference between REGN-EB3 and ansuvimab, the survival difference between those two therapies and ZMapp was significant. This led to the DSMB halting the study and PALM investigators dropping the remdesivir and ZMapp arms from the clinical trial. All patients in the outbreak who elect to participate in the trial will now be given either ansuvimab or REGN-EB3.[21][22][13][12]
On December 21, 2020, the US Food and Drug Administration approved Ebanga (ansuvimab-zykl) for the treatment for Zaire ebolavirus (Ebolavirus) infection in adults and children. Ebanga had been granted US Orphan Drug designation and Breakthrough Therapy designations. Ansuvimab is a human IgG1 monoclonal antibody that binds and neutralizes the virus.
The safety and efficacy of Ebanga were evaluated in the multi-center, open-label, randomized controlled PALM trial. In this study, 174 participants (120 adults and 54 pediatric patients) with confirmed Ebolavirus infection received Ebanga intravenously as a single 50 mg/kg infusion and 168 participants (135 adults and 33 pediatric patients) received an investigational control. The primary efficacy endpoint was 28-day mortality. Of the 174 patients who received Ebanga, 35.1% died after 28 days, compared to 49.4% of the 168 patients who received a control.
Ebanga is the 12th antibody therapeutic to be granted a first approval in the US or EU during 2020.
^ Jump up to:abcdef Clinical trial number NCT03478891 for “Safety and Pharmacokinetics of a Human Monoclonal Antibody, VRC-EBOMAB092-00-AB (MAb114), Administered Intravenously to Healthy Adults” at ClinicalTrials.gov
Literature: Glossop, Paul A.; Watson, Christine A. L.; Price, David A.; Bunnage, Mark E.; Middleton, Donald S.; Wood, Anthony; James, Kim; Roberts, Dannielle; Strang, Ross S.; Yeadon, Michael; Perros-Huguet, Christelle; Clarke, Nicholas P.; Trevethick, Michael A.; MacHin, Ian; Stuart, Emilio F.; Evans, Steven M.; Harrison, Anthony C.; Fairman, David A.; Agoram, Balaji; Burrows, Jane L.; Feeder, Neil; Fulton, Craig K.; Dillon, Barry R.; Entwistle, David A.; Spence, Fiona J. Journal of Medicinal Chemistry, 2011 , vol. 54, # 19 p. 6888 – 6904
PAPER
Organic Process Research & Development (2012), 16(2), 195-203.
An efficient and scalable process for the synthesis of muscarinic antagonist, PF-3635659 1, is described, illustrating redesign of an analogue-targeted synthesis which contained a scale-limiting rhodium-activated C–H amination step. The final route includes a reproducible modified Bouveault reaction which has not previously been reported on a substrate of this complexity, or on such a scale with over 5 kg of the requisite gem-dimethylamine prepared via this methodology.
To a solution of 5-methyl-2,2-diphenyl-5-{3-[3-(prop-2-en-1-yloxy)phenoxy]azetidin1-yl}hexane nitrile 9 (2.8 g, 6.01 mmol) in 3-methyl-pentan-3-ol (30 mL) was added potassium hydroxide (6.7 g, 120 mmol) and the resulting solution was stirred at 120 ºC for 22 hours. The reaction was cooled to room temperature and concentrated in vacuo. The residue was partitioned between ethyl acetate (100 mL) and water (50 mL). The aqueous layer was re-extracted with ethyl acetate (2 x 50 mL). The combined organic layers were dried with MgSO4 and concentrated in vacuo to yield 5-methyl-2,2-diphenyl-5-(3-{3- (propenyl)oxy-phenoxy}-azetidin-1-yl)-hexanamide 10 as a yellow oil (3 g, 6.01 mmol, 100%) which was taken on crude to the next step. To a solution of 5-methyl-2,2-diphenyl-5-(3-{3-(propenyl)oxy-phenoxy}-azetidin-1-yl)- hexanoic acid amide 10 (3.0 g, 6.01 mmol) in methanol (100 mL) was added a 2M aqueous hydrochloric acid solution (30 mL, 15 mmol) and the resulting solution was stirred at 60 ºC for 40 minutes. The volatile solvents were removed in vacuo and the remaining aqueous residue was basified with a saturated aqueous sodium hydrogen carbonate solution. The aqueous layer was extracted with ethyl acetate (3 x 100 mL) and the combined organic layers were dried with magnesium sulphate and concentrated in vacuo.
The crude residue was purified by flash chromatography eluting in ethyl acetate:methanol:ammonia (90:10:1) / pentane (50/50) to yield the title compound 1 as a colourless foam (1.5 g, 3.37 mmol, 54.5%).
Second Discovery Route.
To a solution of 5-[3-(3-methoxyphenoxy)azetidin-1-yl]-5-methyl-2,2-diphenylhexanamide 19 (9.0 g, 19.6 mmol) in dichloromethane (1.25 L) at 0 ºC was dropwise added a solution of boron tribromide (1M in dichloromethane, 58.9 mL, 58.9 mmol) and the mixture stirred for 2 hours at 0 ºC to 20 oC. The mixture was cooled to 0 ºC and quenched with 1M aqueous sodium hydroxide solution (200 mL). The reaction mixture was allowed to warm to 20 oC and stirred as such for 1 hour. The layers were separated and the aqueous layer was extracted with ethyl acetate (2 x 200 mL). The combined organic layers were dried with sodium sulphate and concentrated in vacuo. The crude residue was purified by column chromatography eluting in ethyl acetate:methanol:ammonia (90:10:1) / pentane (50/50) to yield the title compound 1 as a white foam (3.4 g, 7.64 mmol, 39%)
A novel tertiary amine series of potent muscarinic M3 receptor antagonists are described that exhibit potential as inhaled long-acting bronchodilators for the treatment of chronic obstructive pulmonary disease. Geminal dimethyl functionality present in this series of compounds confers very long dissociative half-life (slow off-rate) from the M3 receptor that mediates very long-lasting smooth muscle relaxation in guinea pig tracheal strips. Optimization of pharmacokinetic properties was achieved by combining rapid oxidative clearance with targeted introduction of a phenolic moiety to secure rapid glucuronidation. Together, these attributes minimize systemic exposure following inhalation, mitigate potential drug–drug interactions, and reduce systemically mediated adverse events. Compound 47 (PF-3635659) is identified as a Phase II clinical candidate from this series with in vivo duration of action studies confirming its potential for once-daily use in humans.
Methods and intermediates for preparing the hydrochloride salt of PF-3635659 ,
Cholinergic muscarinic receptors are members of the G-protein coupled receptor super-family and are further divided into 5 subtypes, M to Ms. Muscarinic receptor sub-types are widely and differentially expressed in the body. Genes have been cloned for all 5 sub-types and of these, Mi, M>, and Ms receptors have been extensively pharmacologically characterized in animal and human tissue. Mi receptors are expressed in the brain (cortex and hippocampus), glands and in the ganglia of sympathetic and parasympathetic nerves. M2 receptors are expressed in the heart, hindbrain, smooth muscle and in the synapses of the autonomi c nervous system. Ms receptors are expressed m the brain, glands and smooth muscle. In the airways, stimulation of Ms receptors evokes contraction of airway smooth muscle leading to bronchoeonstnction, while in the salivary-gland Ms receptor stimulation increases fluid and mucus secretion leading to increased salivation. M2 receptors expressed on smooth muscle are understood to be pro-contractile while pre-synaptic M2 receptors modulate acetylcholine release from parasympathetic nerves. Stimulation of M2 receptors expressed in the heart produces bradycardia.
[0003] Short and long-acting muscarinic antagonists are used in the management of asthma and chronic obstructive pulmonary disease (COPD); these include the short acting agents Atrovent® (ipratropium bromide) and Oxivent® (oxitropium bromide) and the long acting agent Spiriva® (tiotropium bromide). These compounds produce bronchodilation following inhaled administration. In addition to improvements in spirometric values, anti-muscarinic use in COPD is associated with improvements m health status and quality of life scores. As a consequence of the wide distribution of muscarinic receptors in the body, significant systemic exposure to muscarinic antagonists is associated with effects such as dry mouth, constipation, mydriasis, urinary retention (all predominantly mediated via blockade of M3 receptors) and tachycardia (mediated by blockade of M2 receptors).
[0004] A newer M3 receptor antagonist that is in the carboxamide family is 5-[3-(3-hydroxyphenoxy)azetidin-l-yl]-5-methyl-2,2-diphenylhexanamide hydrochloride. This carboxamide compound exhibits the following structure (formula II):
[0005] To date, it has not been appreciated that 5-[3-(3-hydroxyphenoxy)azetidin-l-yl]-5-methyl-2,2-diphenylhexanamide hydrochloride can be synthesized from the benzoate salt of 5-[3-(3-hydroxyphenoxy)azetidin~l~y!]-5-methyl-2,2-diphenylhexanenitrile Therefore, there is a need for methods and intermediates used to efficiently prepare 5-[3-(3-hydroxyphenoxy)azetidin~l~y!]-5-methyl-2,2-diphenylhexanamide hydrochloride of good quality from the benzoate salt of 5~[3~ (3~hydroxyphenoxy)azetidin-l-yl]-5-rn ethyl-2, 2-diphenylhexanenitrile.
Reaction Scheme 1 -Preparation of Crude Carboxamide Hydrochloride
formula I formula II
[0061] The coupled benzoate compound of formula 1 can be reacted with KOH, 2-methyl-2-butano!, water, then HC1 aqueous, HC1, and TBME to obtain the crude carboxamide hydrochloride of formula II. The benzoate salt of the nitrile provides for easier purification of the nitrile.
[0062] The reagents useful in the preparation of 5-[3-(3-hydroxyphenoxy)azetidin-l-yl]-5-metiiyl-2,2-diphenyl-hexanamide hydrochloride include a base and an alcohol In some embodiments, a useful base includes potassium hydroxide, while a useful alcohol includes tertiary amyl alcohol also known as 2-methyl-2-butanol. The reaction of the benzoate compound of formula II in tertiary amyl alcohol and potassium hydroxide can be carried in a temperature range from about 85 ± 5°C to about 103 ± 2°C. In a later stage, the temperature of 103 ± 2°C can be maintained in that range for from about 30 hours to about 65 hours. A cooling period to about room temperature is followed by adjusting the pH to a range from about 6.5 to about 8.0. Hydrochloric acid is added to the product of this initial reaction to form a crude carboxamide hydrochloride compound of formula II. The initially isolated crude carboxamide hydrochloride compound of formula II can be washed with an alcohol and then washed with, or slurried in an ether. In some embodiments, the alcohol can be tertiary amyl alcohol and the ether can be methyl tertiary butyl ether.
[0063] In various embodiments, the crude 5-[3-(3-hydroxyphenoxy)azetidin-l-yl]-5-methyl-2,2-diphenylhexanamide hydrochloride can be further purified by treating this carboxamide hydrochloride compound with a slurry of activated charcoal, for example, commercially available ENQPC, PF133 or PF511 SPL (A) carbon, in isopropyl alcohol and water at 85 ± 5°C and filtering as illustrated m the Reaction Scheme 2 below:
Reaction Scheme 2 – Purification of Carboxamide Hydrochloride
Reaction Scheme 3 – Preparation of the Coupled Compound Benzoate
O
[0065] In some embodiments, the benzyl coupled compound of formula III is prepared by reacting an azetidine mesyl HC1 1 -(5-cyano-2-methyl-5,5-diphenylpentan-2-yl)azetidin-3-yl methanes ulfonate hydrochloride with a reagent comprising benzyl resorcinol as illustrated in the Reaction Scheme 4 below:
Reaction Scheme 4 – Preparation of the Benzyl Coupled Compound
In Reaction Scheme 4, the azetidine mesyl hydrochloride of formula IV
is reacted with benzyl resorcinol of formula V
The reagent can comprise benzyl resorcinol and, in some aspects, acetonitrile, a carbonate salt of either cesium or potassium, sodium hydroxide, water, ethyl acetate, hexanes or a mixture thereof. The order of addition of reagents in this step overcomes the need for specific equipment (e.g., a bespoke/unusual agitator) and allows the step to be run in a general purpose reactor.
[0066] Benzyl resorcinol is commercially available and can be obtained commercially, for example, from Sigma Aldrich Corp. In various embodiments, benzyl resorcinol of formula V can be prepared by reacting resorcinol with benzyl chloride to form benzyl resorcinol according to the Reaction Scheme 5 below:
Reaction Scheme 5 — Preparation of Benzyl Resorcinol
Resorcinol DMF/Hexane
Toluene Benzyl Resorcinol
or
3-{benzyioxy) phenol
V
[0067] In certain aspects, the benzyl resorcinol is prepared by reacting resorcinol with benzyl chloride m a reagent which can include potassium carbonate, dimethylformamide, water, sodium hydroxide, toluene, hydrochloric acid, hexanes or a combination thereof. In some instances, benzyl resorcinol seeding material may also be added. For the conversion of the resorcinol to the benzyl resorcinol (V), the developed chemistry’- allows effective removal of remaining resorcinol starting material and dibenzyl impurity to give the benzyl resorcinol product in good yield and quality.
Reaction Scheme 6 – Preparation of Azetidine Mesyl Hydrochloride
Azetidine alcohol Azetidine mesyl
VI hydrochloride
Reaction Scheme 7 – Preparation of Azetidine Alcohol
Scheme 8 – Preparation of Diphenyl Amine
Reaction Scheme 9 Preparation of Diphenyl Chloro Amide
Reaction Scheme 10 – Preparation of Diphenyl Alkene
The compound was originally claimed without an action as example 108 in WO2007034325 , for the treatment of chronic obstructive pulmonary disease, and this is the first filing from Pfizer relating to the compound since the program was presumed discontinued in 2011.
Example 108 5-r3-(3-Hvdroxyphenoxy)azetidin-1-vπ-5-methyl-2,2-diphenylhexanamide
Boron tribromide (1M in dichloromethane, 1.75mL, 1.75mmol) was added to an ice-cooled solution of the product of example 100 (200mg, 0.44mmol) in dichloromethane (5mL) and the mixture was stirred at O0C for 1 hour. Further boron tribromide (1M in dichloromethane, 0.5mL, O.δmmol) was added and the mixture was stirred at O0C for 30 minutes. The reaction was then quenched with 1M sodium hydroxide solution (5mL), diluted with dichloromethane (2OmL) and stirred at room temperature for 40 minutes. The aqueous layer was separated, extracted with ethyl acetate (2x25mL) and the combined organic solution was dried over magnesium sulfate and concentrated in vacuo. Purification of the residue by column chromatography on silica gel, eluting with pentane:ethyl acetate/methanol/0.88 ammonia (90/10/1), 75:25 to 50:50, afforded the title compound as a colourless foam in 91% yield, 176mg.
It does however, follow on from WO2018167804 , assigned solely to Mylan , claiming amorphous and crystalline forms designated as Forms I-XI, for treating allergy, and this seems to confirm the potential of the candidate is being revisited, and possibly licensed.
(5-[3-(3-Hydroxyphenoxy)azetidin-l-yl]-5-methyl-2,2-diphenylhexanamide hydrochloride has a structure depicted below as Compound-A.
Compound-A
Compound-A is a muscarinic antagonist useful for treating allergy or respiratory chronic obstructive pulmonary disease.
Compound-A and pharmaceutically acceptable salts are claimed in U.S. Pat. No. 7,772,223 B2 and one of its non-solvated crystalline forms is claimed in U.S. Pat. No. 8,263,583 B2.
Examples:
Example 1: Processes for the preparation of amorphous form of Compound-A.
Compound-A (5 g) was dissolved in methanol (150 ml) at 60-65°C. The solution was filtered at 60-65°C to remove undissolved particulate and then cooled to 25-30°C. The clear solution of Compound-A was subjected to spray drying in a laboratory Spray Dryer (Model Buchi-290) with a 5 ml/min feed rate of the solution and inlet temperature at 75°C with 100% aspiration to yield an amorphous form of Compound-A.
Puldysa (idebenone), for the treatment of Duchenne muscular dystrophyTitle: Idebenone CAS Registry Number: 58186-27-9 CAS Name: 2-(10-Hydroxydecyl)-5,6-dimethoxy-3-methyl-2,5-cyclohexadiene-1,4-dione Additional Names: 6-(10-hydroxydecyl)-2,3-dimethoxy-5-methyl-1,4-benzoquinone; 2,3-dimethoxy-5-methyl-6-(10¢-hydroxydecyl)-1,4-benzoquinone; 6-(10-hydroxydecyl)ubiquinone Manufacturers’ Codes: CV-2619 Trademarks: Avan (Takeda); Daruma (Takeda); Lucebanol (Hormona); Mnesis (Takeda) Molecular Formula: C19H30O5Molecular Weight: 338.44 Percent Composition: C 67.43%, H 8.93%, O 23.64% Literature References: Ubiquinone derivative with protective effects against cerebral ischemia. Prepn: H. Morimoto et al.,DE2519730; eidem,US4271083 (1975, 1981 both to Takeda); K. Okamoto et al.,Chem. Pharm. Bull.30, 2797 (1982); C.-A. Yu, L. Yu, Biochemistry21, 4096 (1982). Effect on ischemia-induced amnesia in rats: N. Yamazaki et al.,Jpn. J. Pharmacol.36, 349 (1984). Metabolism in animals: T. Kobayashi et al.,J. Pharmacobio-Dyn.8, 448 (1985). Disposition: H. Torii et al.,ibid. 457. Pharmacokinetics and tolerance in humans: M. F. Barkworth et al.,Arzneim.-Forsch.35, 1704 (1985). Series of articles on pharmacology and clinical studies: Arch. Gerontol. Geriatr.8, 193-366 (1989). Review of chemistry, toxicology and pharmacology: I. Zs-Nagy, Arch. Gerontol. Geriatr.11, 177-186 (1990).Properties: Orange needles from ligroin, mp 46-50° (Morimoto); also reported as crystals from hexane + ethyl acetate, mp 52-53° (Okamoto). Sol in organic solvents. Practically insol in water.Melting point: mp 46-50° (Morimoto); mp 52-53° (Okamoto)Therap-Cat: Nootropic.Keywords: Nootropic.
Idebenone is a member of the class of 1,4-benzoquinones which is substituted by methoxy groups at positions 2 and 3, by a methyl group at positions 5, and by a 10-hydroxydecyl group at positions 6. Initially developed for the treatment of Alzheimer’s disease, benefits were modest; it was subsequently found to be of benefit for the symptomatic treatment of Friedreich’s ataxia. It has a role as an antioxidant. It is a primary alcohol and a member of 1,4-benzoquinones.
Research on idebenone as a potential therapy of Alzheimer’s disease have been inconsistent, but there may be a trend for a slight benefit.[7][8] In May 1998, the approval for this indication was cancelled in Japan due to the lack of proven effects. In some European countries, the drug is available for the treatment of individual patients in special cases.[1]
Friedreich’s ataxia (Sovrima)
Preliminary testing has been done in humans and found idebenone to be a safe treatment for Friedreich’s ataxia (FA), exhibiting a positive effect on cardiac hypertrophy and neurological function.[9] The latter was only significantly improved in young patients.[10] In a different experiment, a one-year test on eight patients, idebenone reduced the rate of deterioration of cardiac function, but without halting the progression of ataxia.[11]
The drug was approved for FA in Canada in 2008 under conditions including proof of efficacy in further clinical trials.[12] However, on February 27, 2013, Health Canada announced that idebenone would be voluntarily recalled as of April 30, 2013 by its Canadian manufacturer, Santhera Pharmaceuticals, due to the failure of the drug to show efficacy in the further clinical trials that were conducted.[13] In 2008, the European Medicines Agency (EMA) refused a marketing authorisation for this indication.[1] As of 2013 the drug was not approved for FA in Europe[14] nor in the US, where there is no approved treatment.[15]
Leber’s hereditary optic neuropathy (Raxone)
Leber’s hereditary optic neuropathy (LHON) is a mitochondrially inherited (mother to all offspring) degeneration of retinal ganglion cells (RGCs) and their axons that leads to an acute or subacute loss of central vision; this affects predominantly young adult males. Santhera completed a Phase III clinical trial in this indication in Europe with positive results,[16] and submitted an application to market the drug to European regulators in July 2011.[17] It is approved by EMA for this indication and was designated an orphan drug in 2007.[4]
Indications being explored
Duchenne muscular dystrophy (Catena)
After experiments in mice[18] and preliminary studies in humans, idebenone has entered Phase II clinical trials in 2005[3] and Phase III trials in 2009.[19]
Other neuromuscular diseases
Phase I and II clinical trials for the treatment of MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes)[20] and primary progressive multiple sclerosis[21] are ongoing as of December 2013.
Life style
Idebenone is claimed to have properties similar to CoQ10 in its antioxidant properties, and has therefore been used in anti-aging on the basis of free-radical theory. Clinical evidence for this use is missing. It has been used in topical applications to treat wrinkles.[22]
Pharmacology
In cellular and tissue models, idebenone acts as a transporter in the electron transport chain of mitochondria and thus increases the production of adenosine triphosphate (ATP) which is the main energy source for cells, and also inhibits lipoperoxide formation. Positive effects on the energy household of mitochondria has also been observed in animal models.[1][23] Clinical relevance of these findings has not been established.
Pharmacokinetics
Idebenone is well absorbed from the gut but undergoes excessive first pass metabolism in the liver, so that less than 1% reach the circulation. This rate can be improved with special formulations (suspensions) of idebenone and by administering it together with fat food; but even taking these measures bioavailability still seems to be considerably less than 14% in humans. More than 99% of the circulating drug are bound to plasma proteins. Idebenone metabolites include glucuronides and sulfates, which are mainly (~80%) excreted via the urine.[1]
The palladium-catalyzed olefination of a sp2 or benzylic carbon attached to a (pseudo)halogen is known as the Heck reaction.2,63 It is a powerful tool, mainly used for the synthesis of vinylarenes, and it has also been employed for the construction of conjugated double bonds. The widespread application of this reaction can be illustrated by numerous examples in both academia small-scale64 and industrial syntheses.5 As an example, in 2011, a idebenone (124) total synthesis based on a Heck reaction was described (Scheme 35).65 This compound, initially designed for the treatment of Alzheimer’s and Parkinson’s diseases, presented a plethora of other interesting activities, such as free radical scavenging and action against some muscular illnesses. The key step in the synthesis was the coupling of 2-bromo-3,4,5-trimethoxy-1-methylbenzene (125) with dec-9-en-1-ol affording products 126. Under non-optimized conditions (Pd(OAc)2, PPh3, Et3N, 120 ºC), a mixture composed of 60% linear olefins 126 and 15% of the undesired branched product 127 was obtained after three days of reaction. Therefore, the conditions were optimized, allowing the preparation of 126 in 67% yield with no detection of 127 after only 30 min of reaction employing DMF, Pd(PPh3)4, iPr2NEt under microwave heating. To conclude the synthesis, the Heck adducts were submitted to hydroxyl protection/deprotection, hydrogenation, and ring oxidation. After these reactions, idebenone was obtained with 20% overall yield over 6 steps.
Scheme 35 Synthesis of idebenone (124) based on Heck reaction of 2-bromo-3,4,5-trimethoxy-1-methylbenzene with dec-9-en-1-ol under microwave irradiation.
An environmentally benign, convenient, high yielding, and cost-effective synthesis leading to idebenone is disclosed. The synthesis includes a bromination process for the preparation of 2-bromo-3,4,5-trimethoxy-1-methylbenzene, a protocol for the Heck cross-coupling reaction using either thermal or microwave heating, olefin reduction by palladium catalyzed hydrogenation, and a green oxidation protocol with hydrogen peroxide as oxidant to achieve the benzoquinone framework. The total synthesis is composed of six steps that provide an overall yield of 20% that corresponds to a step yield of 76%.
Analogues of mitoQ and idebenone were synthesized to define the structural elements that support oxygen consumption in the mitochondrial respiratory chain. Eight analogues were prepared and fully characterized, then evaluated for their ability to support oxygen consumption in the mitochondrial respiratory chain. While oxygen consumption was strongly inhibited by mitoQ analogues 2–4 in a chain length-dependent manner, modification of idebenone by replacement of the quinone methoxy groups by methyl groups (analogues 6–8) reduced, but did not eliminate, oxygen consumption. Idebenone analogues 6–8 also displayed significant cytoprotective properties toward cultured mammalian cells in which glutathione had been depleted by treatment with diethyl maleate.
Idebenone (5)18 To a stirred solution containing 200 mg (0.467 mmol) of 2,3- dimethoxy-6-methyl-5-benzyloxydecyl-p-benzoquinone (38) in 5 mL of anhydrous methanol at 23 C was added 15 mg of 10 % Pd/C in one portion. The reaction mixture was stirred at 23 C under an atmosphere of hydrogen for 24 h. Air was then bubbled through the reaction mixture at 23 C for 24 h. The suspension was filtered through Celite and the filtrate was concentrated under diminished pressure to afford idebenone (5) as an orange solid: yield 130 mg (82%); mp: 46–47 C; 1 H NMR (400 MHz, CDCl3) d 1.34 (m, 14H), 1.60 (quint, 2H, J = 7.6 Hz), 2.04 (s, 3H), 2.44 (t, 2H, J = 8.0 Hz), 3.63 (t, 2H, J = 6.8 Hz), and 3.99 (s, 6H); 13C NMR (100 MHz, CDCl3) d 11.9, 25.7, 26.4, 28.7, 29.3, 29.3, 29.4, 29.5, 29.8, 32.7
^ Jump up to:ab Clinical trial number NCT00654784 for “Efficacy and Tolerability of Idebenone in Boys With Cardiac Dysfunction Associated With Duchenne Muscular Dystrophy (DELPHI)” at ClinicalTrials.gov
^ Liu, XJ; Wu, WT (1999). “Effects of ligustrazine, tanshinone II A, ubiquinone, and idebenone on mouse water maze performance”. Zhongguo Yao Li Xue Bao. 20 (11): 987–90. PMID11270979.
^ Schaffler, K; Hadler, D; Stark, M (1998). “Dose-effect relationship of idebenone in an experimental cerebral deficit model. Pilot study in healthy young volunteers with piracetam as reference drug”. Arzneimittel-Forschung. 48 (7): 720–6. PMID9706371.
^ Gutzmann, H; Kühl, KP; Hadler, D; Rapp, MA (2002). “Safety and efficacy of idebenone versus tacrine in patients with Alzheimer’s disease: results of a randomized, double-blind, parallel-group multicenter study”. Pharmacopsychiatry. 35 (1): 12–8. doi:10.1055/s-2002-19833. PMID11819153.
^ Clinical trial number NCT01027884 for “Phase III Study of Idebenone in Duchenne Muscular Dystrophy (DMD) (DELOS)” at ClinicalTrials.gov
^ Clinical trial number NCT00887562 for “Study of Idebenone in the Treatment of Mitochondrial Encephalopathy Lactic Acidosis & Stroke-like Episodes (MELAS)” at ClinicalTrials.gov
^ Clinical trial number NCT00950248 for “Double Blind Placebo-Controlled Phase I/II Clinical Trial of Idebenone in Patients With Primary Progressive Multiple Sclerosis (IPPoMS)” at ClinicalTrials.gov
^ McDaniel D, Neudecker B, Dinardo J, Lewis J, Maibach H (September 2005). “Clinical efficacy assessment in photodamaged skin of 0.5% and 1.0% idebenone”. J Cosmet Dermatol. 4 (3): 167–73. doi:10.1111/j.1473-2165.2005.00305.x. PMID17129261. S2CID2394666.
Inclisiran was first developed by Alnylam Pharmaceuticals, Inc. (Cambridge, Massachusetts, US). Development has now been assumed by The Medicines Company (Parsippany, New Jersey, US). One phase I and two phase II trials have been completed. Topline results of two phase III trials were also recently presented while other phase III trials are still ongoing as part of the ORION clinical development program. …..https://www.ncbi.nlm.nih.gov/books/NBK555477/
Inclisiran is a long-acting, synthetic small interfering RNA (siRNA) directed against proprotein convertase subtilisin-kexin type 9 (PCSK9), which is a serine protease that regulates plasma low-density lipoprotein cholesterol (LDL-C) levels. By binding to PCSK9 messenger RNA, inclisiran prevents protein translation of PCSK9, leading to decreased concentrations of PCSK9 and plasma concentrations of LDL cholesterol.1,2 Lowering circulating plasma LDL-C levels offers an additional benefit of reducing the risk of cardiovascular disease (CVD) and improving cardiovascular outcomes, as hypercholesterolemia is a major known risk factor for CVD.1,2
On December 11, 2020, the European Commission (EC) granted authorization for marketing inclisiran as the first and only approved siRNA for the treatment of adults with primary hypercholesterolemia (heterozygous familial and non-familial) or mixed dyslipidemia, alone or in combination with other lipid-lowering therapies. It is marketed under the trade name Leqvio 8 and is also currently under review by the FDA.
On 15 October 2020, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency (EMA) adopted a positive opinion, recommending the granting of a marketing authorization for the medicinal product Leqvio, intended for the treatment for primary hypercholesterolaemia or mixed dyslipidaemia.[6] Inclisiran was approved for use in the European Union in December 2020.[1]
History
In 2019 The Medicines Company announced positive results from pivotal phase III study (all primary and secondary endpoints were met with efficacy consistent with Phase I and II studies). The company anticipates regulatory submissions in the U.S. in the fourth quarter of 2019, and in Europe in the first quarter of 2020.[7] The Medicines Company is being acquired by Novartis.[8]
“Inclisiran”. Drug Information Portal. U.S. National Library of Medicine.
Clinical trial number NCT03399370 for “Inclisiran for Participants With Atherosclerotic Cardiovascular Disease and Elevated Low-density Lipoprotein Cholesterol (ORION-10)” at ClinicalTrials.gov
Clinical trial number NCT03400800 for “Inclisiran for Subjects With ACSVD or ACSVD-Risk Equivalents and Elevated Low-density Lipoprotein Cholesterol (ORION-11)” at ClinicalTrials.gov
Kosmas CE, Munoz Estrella A, Sourlas A, Silverio D, Hilario E, Montan PD, Guzman E: Inclisiran: A New Promising Agent in the Management of Hypercholesterolemia. Diseases. 2018 Jul 13;6(3). pii: diseases6030063. doi: 10.3390/diseases6030063. [PubMed:30011788]
German CA, Shapiro MD: Small Interfering RNA Therapeutic Inclisiran: A New Approach to Targeting PCSK9. BioDrugs. 2020 Feb;34(1):1-9. doi: 10.1007/s40259-019-00399-6. [PubMed:31782112]
Doggrell SA: Inclisiran, the billion-dollar drug, to lower LDL cholesterol – is it worth it? Expert Opin Pharmacother. 2020 Nov;21(16):1971-1974. doi: 10.1080/14656566.2020.1799978. Epub 2020 Aug 4. [PubMed:32749892]
Goldstein JL, Brown MS: Regulation of low-density lipoprotein receptors: implications for pathogenesis and therapy of hypercholesterolemia and atherosclerosis. Circulation. 1987 Sep;76(3):504-7. doi: 10.1161/01.cir.76.3.504. [PubMed:3621516]
Leiter LA, Teoh H, Kallend D, Wright RS, Landmesser U, Wijngaard PLJ, Kastelein JJP, Ray KK: Inclisiran Lowers LDL-C and PCSK9 Irrespective of Diabetes Status: The ORION-1 Randomized Clinical Trial. Diabetes Care. 2019 Jan;42(1):173-176. doi: 10.2337/dc18-1491. Epub 2018 Nov 28. [PubMed:30487231]
Cupido AJ, Kastelein JJP: Inclisiran for the treatment of hypercholesterolaemia: implications and unanswered questions from the ORION trials. Cardiovasc Res. 2020 Sep 1;116(11):e136-e139. doi: 10.1093/cvr/cvaa212. [PubMed:32766688]
Novartis: Novartis receives EU approval for Leqvio (inclisiran), a first-in-class siRNA to lower cholesterol with two doses a year [Link]
Summary of Product Characteristics: Leqvio (inclisiran), solution for subcutaneous injection [Link]
Summary
Atherosclerotic cardiovascular disease (ASCVD) remains one of the leading causes of death in Canada. Cholesterol, specifically low-density lipoprotein cholesterol (LDL-C), is a major risk factor for cardiovascular disease (CVD) and is thereby targeted to reduce the likelihood of a cardiovascular event, such as a myocardial infarction (MI) and stroke.
Inclisiran, first developed by Alnylam Pharmaceuticals, Inc. (Cambridge, Massachusetts, US) then by The Medicines Company (Parsippany, New Jersey, US), is a small interfering ribonucleic acid (siRNA) molecule being investigated for the treatment of hypercholesterolemia.
ORION-1 was a phase II, double-blind, placebo-controlled, multi-centre, randomized controlled trial of 501 patients. Patients were included in the trial if they had a history of ASCVD or were at high risk of ASCVD. The treatment arms were administered 200 mg, 300 mg, or 500 mg of inclisiran on day 1, or 100 mg, 200 mg, or 300 mg of inclisiran on days 1 and 90. The comparator was either placebo on day 1 or placebo on days 1 and 90. The primary end point was percentage change in LDL-C at day 180 from baseline.
The ORION-1 study demonstrated that inclisiran, administered at various doses and intervals, compared with placebo, resulted in a statistically significant reduction in LDL-C levels (P < 0.001 for all comparisons versus placebo). The greatest reduction in LDL-C levels was obtained with the 300 mg dose of inclisiran given at days 1 and 90 with a 52.6% (95% confidence interval [CI]: −57.1 to −48.1) reduction at day 180 compared with baseline, and a mean absolute reduction in LDL-C levels of 1.66 (standard deviation 0.54) mmol/L. Results from the ORION-1 trial provided the necessary data to make a decision regarding the dosing regimen to be used in subsequent phase III trials, in particular the ORION-11 phase III trial.
The ORION-11 study was a phase III international, multi-centre, and double-blind trial which randomized 1,617 participants (87% with established ASCVD) to inclisiran 300 mg (n = 810) or placebo (n = 807). An initial inclisiran dose of 300 mg given subcutaneously was administered at day 1, day 90, and then every six months for two doses, that is at days 270 and 450. The mean baseline LDL-C level was 2.8 mmol/L (inclisiran) and 2.7 mmol/L (placebo); 96% of participants were on high-dose statin therapy. There was a 50% time-averaged reduction in LDL-C levels from day 90 to day 540 (P < 0.00001). Pre-specified exploratory cardiovascular composite end point (cardiac death, cardiac arrest, MI, or stroke) occurred in 7.8% of inclisiran treated patients versus 10.3% of patients on placebo; this lower rate was mainly driven by a reduction in MI and stroke. With respect to adverse effects, 4.69% of patients on inclisiran reported an injection site reaction, compared with 0.5% of patients on placebo. All reactions were transient. There was no evidence of liver, kidney, muscle, or platelet toxicity.
Inclisiran may be an option in the future as a cholesterol-lowering medication, where it would likely be used in patients who are unable to achieve their LDL-C targets despite maximally tolerated statin therapy or who are intolerant to statin therapy. However, results from the inclisiran cardiovascular outcome trial (ORION-4), are needed to confirm its efficacy in reducing CVD and its long-term safety.
Inclisiran is not yet approved by any regulatory authority, but its ORION clinical development program identifies the year 2021 as the goal to reach worldwide markets.
(+)-Ketamine(2S)-2-(2-Chlorophenyl)-2-(methylamino)cyclohexanone (S)-Ketamine33643-46-8[RN]7884Cyclohexanone, 2-(2-chlorophenyl)-2-(methylamino)-, (2S)-Cyclohexanone, 2-(2-chlorophenyl)-2-(methylamino)-, (S)- KetamineCAS Registry Number: 6740-88-1CAS Name: 2-(2-Chlorophenyl)-2-(methylamino)cyclohexanoneMolecular Formula: C13H16ClNOMolecular Weight: 237.73Percent Composition: C 65.68%, H 6.78%, Cl 14.91%, N 5.89%, O 6.73%Literature References: Prepn: C. L. Stevens, BE634208; idem,US3254124 (1963, 1966 both to Parke, Davis). Isoln of optical isomers: T. W. Hudyma et al.,DE2062620 (1971 to Bristol-Myers), C.A.75, 118119x (1971). Clinical pharmacology of racemate and enantiomers: P. F. White et al.,Anesthesiology52, 231 (1980). Toxicity: E. J. Goldenthal, Toxicol. Appl. Pharmacol.18, 185 (1971). Enantioselective HPLC determn in plasma: G. Geisslinger et al.,J. Chromatogr.568, 165 (1991). Comprehensive description: W. C. Sass, S. A. Fusari, Anal. Profiles Drug Subs.6, 297-322 (1977). Review of pharmacology and use in veterinary medicine: M. Wright, J. Am. Vet. Med. Assoc.180, 1462-1471 (1982). Review of pharmacology and clinical experience: D. L. Reich, G. Silvay, Can. J. Anaesth.36, 186-197 (1989); in pediatric procedures: S. M. Green, N. E. Johnson, Ann. Emerg. Med.19, 1033-1046 (1990).Properties: Crystals from pentane-ether, mp 92-93°. uv max (0.01N NaOH in 95% methanol): 301, 276, 268, 261 nm (A1%1cm 5.0, 7.0, 9.8, 10.5). pKa 7.5. pH of 10% aq soln 3.5.Melting point: mp 92-93°pKa: pKa 7.5Absorption maximum: uv max (0.01N NaOH in 95% methanol): 301, 276, 268, 261 nm (A1%1cm 5.0, 7.0, 9.8, 10.5) Derivative Type: HydrochlorideCAS Registry Number: 1867-66-9Manufacturers’ Codes: CI-581Trademarks: Ketalar (Pfizer); Ketanest (Pfizer); Ketaset (Fort Dodge); Ketavet (Gellini); Vetalar (Bioniche)Molecular Formula: C13H16ClNO.HClMolecular Weight: 274.19Percent Composition: C 56.95%, H 6.25%, Cl 25.86%, N 5.11%, O 5.84%Properties: White crystals, mp 262-263°. Soly in water: 20 g/100 ml. LD50 in adult mice, rats (mg/kg): 224 ±4, 229 ±5 i.p. (Goldenthal).Melting point: mp 262-263°Toxicity data: LD50 in adult mice, rats (mg/kg): 224 ±4, 229 ±5 i.p. (Goldenthal) NOTE: This is a controlled substance (depressant): 21 CFR, 1308.13.Therap-Cat: Anesthetic (intravenous).Therap-Cat-Vet: Anesthetic (intravenous).Keywords: Anesthetic (Intravenous).Esketamine hydrochloride, S enantiomer of ketamine, is in phase III clinical trials by Johnson & Johnson for the treatment of depression.Drug Name:Esketamine HydrochlorideResearchCode:JNJ-54135419MOA:Dopamine reuptake inhibitor; NMDA receptor antagonistIndication:DepressionStatus:Phase III (Active)Company:Johnson & Johnson (Originator)
50 g (0.21 mol) R,S-ketamine are dissolved in 613 ml of acetone at the boiling point and subsequently mixed with 31.5 g (0.21 mol) L-(+)-tartaric acid. In order to obtain a clear solution, 40 ml of water are added thereto at the boiling point and subsequently the clear solution is filtered off while still hot. After the addition of seed crystals obtained in a small preliminary experiment, the whole is allowed to cool to ambient temperature while stirring. After standing overnight, the crystals formed are filtered off with suction and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.).
Yield (tartrate): 64.8 g
m.p.: 161° C.
[α]D : +26.1° (c=2/H2 O)
Thereafter, the crystallisate is recrystallised in a mixture of 1226 ml acetone and 90 ml water. After cooling to ambient temperature and subsequently stirring for 4 hours, the crystals are filtered off with suction and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C). There are obtained 38.8 g of tartrate (95.29% of theory).
m.p.: 175.3° C.
[α]D : +68.9° (c=2/H2 O)
The base is liberated by taking up 38.8 g of tartrate in 420 ml of aqueous sodium hydroxide solution and stirring with 540 ml of diethyl ether. The ethereal phase is first washed with water and subsequently with a saturated solution of sodium chloride. The organic phase is dried over anhydrous sodium sulphate. After filtering, the solution is evaporated to dryness on a rotary evaporator, a crystalline, colourless product remaining behind.
In order possibly to achieve a further purification, the base can be recrystallised from cyclohexane. For this purpose, 10.75 g of the crude base are dissolved in 43 ml cyclohexane at the boiling point. While stirring, the clear solution is slowly cooled to about 10° C. and then stirred at this temperature for about 1 hour. The crystallisate which precipitates out is filtered off with suction and dried to constant weight.
125 ml of water are taken and subsequently 31.5 g (0.21 mol) L-(+)-tartaric acid and 50 g (0.21 mol) R,S-ketamine added thereto. While stirring, this mixture is warmed to 50-60° C. until a clear solution results. After cooling to ambient temperature while stirring and subsequently stirring overnight, the crystals formed are filtered off with suction. Subsequently, the crystallisate is first washed with water (1-6° C.) and subsequently washed twice with, in each case, 20 ml of acetone. Drying in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.) gives 31.79 g of tartrate (78.23%) of theory).
EXAMPLE 3
150 ml of water are taken and subsequently mixed with 39.8 g (0.27 mol) L-(+)-tartaric acid and 50 g (0.21 mol) R,S-ketamine. While stirring, this mixture is warmed to 50-60° C. until a clear solution results.
After cooling to ambient temperature while stirring and subsequently stirring overnight, the crystals formed are filtered off with suction. Subsequently, the crystallisate is successively washed with 8 ml of water (1-6° C.) and thereafter twice with, in each case, 20 ml acetone.
Drying in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.) gives 32.58 g of tartrate (80.02% of theory).
EXAMPLE 4
150 ml of water and 50 ml isopropanol are taken. After the addition of 39.8 g (0.21 mol) L-(+)-tartaric acid and 50 g (0.21 mol) R,S-ketamine, the mixture is heated to reflux temperature while stirring until a solution results (possibly add water until all is dissolved).
Subsequently, while stirring, the solution is allowed to cool to ambient temperature and stirred overnight. The crystals are filtered off with suction and subsequently washed with a 1:2 mixture of 20 ml of water/isopropanol and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.). There are obtained 24.45 g of tartrate (62.63% of theory).
EXAMPLE 5
50 g (0.21 mol) R,S-ketamine are dissolved at the boiling point in 300 ml acetone and subsequently mixed with 31.5 g (0.21 mol) L-(+)-tartaric acid and 100 ml of water. The whole is allowed to cool while stirring and possibly seeded.
After standing overnight, the crystals formed are filtered off with suction, then washed twice with, in each case, 20 ml acetone and dried in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.). There are obtained 30.30 g of tartrate (74.57% of theory).
EXAMPLE 6
75 ml of water and 50 ml isopropanol are taken and subsequently 39.8 g (0.27 mol) L-(+)-tartaric acid added thereto. While stirring, the mixture is heated to reflux temperature until a clear solution results. After cooling to ambient temperature while stirring and subsequently stirring overnight, the crystals formed are filtered off with suction. Subsequently, the crystallisate is washed with a 1:2 mixture of 20 ml water/isopropanol. After drying in a circulating air drying cabinet (first at ambient temperature and then at 50-60° C.), there are obtained 34.84 g of tartrate (85.74% of theory).
EXAMPLE 7
20 g of the S-(+)-tartrate obtained in Example 4 are dissolved in 100 ml of water at 30-40° C. With about 7 ml of 50% sodium hydroxide solution, an S-(-)-ketamine base is precipitated out up to about pH 13. It is filtered off with suction and washed neutral with water to pH 7-8. Subsequently, it is dried for about 24 hours at 50° C. in a circulating air drying cabinet. There are obtained 11.93 g S-(-)-ketamine (97.79% of theory).
EXAMPLE 8
5 g of the S-(-)-ketamine obtained in Example 7 are dissolved in 50 ml isopropanol at about 50° C. and possibly filtered off with suction over kieselguhr. Subsequently, gaseous hydrogen chloride is passed in at 50-60° C. until a pH value of 0-1 is reached. The reaction mixture is allowed to cool to ambient temperature, filtered off with suction and washed with about 5 ml isopropanol. The moist product is dried overnight at about 50° C. in a circulating air drying cabinet. There are obtained 5.09 g S-(+)-ketamine hydrochloride (88.06% of theory).
Route 2
Reference:1. J. Am. Chem. Soc.2015, 137, 3205-3208.
Here we report the direct asymmetric amination of α-substituted cyclic ketones catalyzed by a chiral phosphoric acid, yielding products with a N-containing quaternary stereocenter in high yields and excellent enantioselectivities. Kinetic resolution of the starting ketone was also found to occur on some of the substrates under milder conditions, providing enantioenriched α-branched ketones, another important building block in organic synthesis. The utility of this methodology was demonstrated in the short synthesis of (S)-ketamine, the more active enantiomer of this versatile pharmaceutical.
CLIP
Initial reagent: cyclopentyl Grignard Step 0: Producing cyclopentyl Grignard Reacting cyclopentyl bromide with magnesium in solvent (ether or THF) Best results: distill solvent from Grignard under vacuum and replace with hydrocarbon solvent (e.g. benzene) Step 1: processing to (o-chlorophenyl)-cyclopentyl ketone Adding o-chlorobenzonitrile to cyclopentyl Grignard in solvent, stirring for long period of time (typically three days) Hydrolyzing reaction with mixture containing crushed ice, ammonium chloride and some ammonium hydroxide Extraction with organic solvent gives (o-chlorophenyl)-cyclopentyl ketone
Step 2: processing to alpha-bromo (o-chlorophenyl)-cyclopentyl ketone ketone processed with bromine in carbon tetrachloride at low temperature (typical T = 0°C), addition of bromine dropwise forming orange suspension Suspension washed in dilute aquerous solution of sodium bisufide and evaporated giving 1-bromocyclopentyl-(o-chlorophenyl)-ketone Note: bromoketone is unstable, immeadiate usage. Bromination carried out with NBromosuccinimide result higher yield (roughly 77%) Step 3: processing to 1-hydroxycyclopentyl-(o-chlorophenyl)-ketone-N-methylimine Dissolving bromoketone in liquid methylamine freebase (or benzene as possible solvent) After time lapse (1h): excess methylamine evaporated, residue dissolved in pentane and filtered evaporation of solvent yields 1-hydroxy-cyclopentyl-(o-chlorophenyl)-ketone N-methylimine Note: longer time span (4-5d) for evaporation of methylaminemay increase yield Step 4: processing to 2-Methylamino-2-(o-chlorophenyl)-cyclohexanone (Ketamine) Method: Thermal rearragement (qualitative yield after 30min in 180°C) N-methylimine dissolved in 15ml decalin, refluxed for 2.5h Evaporation of solvent under reduced temperature followed by extraction of residue with dilute hydrochloric acid Treatment with decolorizing charcoal (solution: acidic => basic) Recrystallization from pentane-ether Note – alternative to use of decalin: pressure bomb
racemic compound, in pharmaceutical preparation racemic more active enantiomere esketamine (S-Ketamine) available as Ketanest S, but Arketamine (R-Ketamine) never marketed for clinical use, Optical rotation: varies between salt and free base form free base form: (S)-Ketamine dextrorotation (S)-(+)-ketamine hydrochloridesalt: levorotation(S)-(-)-ketamine Reason found in molecular level: different orientation of substituents: freebase: o-chlorophenyl equatorial, methylamino axia
Process Research and Impurity Control Strategy of Esketamine Organic Process Research & Development ( IF 3.023 ) Pub Date: 2020-03-18 , DOI: 10.1021/acs.oprd.9b00553 Shenghua Gao; Xuezhi Gao; Zhezhou Yang; Fuli Zhang An improved synthesis of ( S )-ketamine (esketamine) has been developed, which was cost-effective, and the undesired isomer could be recovered by racemization. Critical process parameters of each step were identified as well as the process-related impurities. The formation mechanisms and control strategies of most impurities were first discussed. Moreover, the ( S )-ketamine tartrate is a dihydrate, which was disclosed for the first time. The practicable racemization catalyzed by aluminum chloride was carried out in quantitative yield with 99% purity . The ICH-grade quality ( S)-ketamine hydrochloride was obtained in 51.1% overall yield (14.0% without racemization) by chiral resolution with three times recycling of the mother liquors. The robust process of esketamine could be industrially scalable.
Process Research and ketamine impurity control strategy
has been developed an improved ( S ) – ketamine (esketamine) synthesis, the high cost-effective way, the undesired isomer may be recycled by racemization. Determine the key process parameters and process-related impurities for each step. First, the formation mechanism and control strategy of most impurities are discussed. In addition, ( S )-ketamine tartrate is a dihydrate, which is the first time it has been published. The feasible racemization catalyzed by aluminum chloride proceeds in a quantitative yield with a purity of 99%. ICH grade quality ( S) 5-ketamine hydrochloride can be obtained through chiral resolution and three times the mother liquor recovery rate. The total yield is 51.1% (14.0% without racemization). The robust process of ketamine can be used in Industrial promotion.
Taghizadeh, M.J., Gohari, S.J.A., Javidan, A. et al. A novel strategy for the asymmetric synthesis of (S)-ketamine using (S)-tert-butanesulfinamide and 1,2-cyclohexanedione. J IRAN CHEM SOC15, 2175–2181 (2018). https://doi.org/10.1007/s13738-018-1404-1
We present a novel asymmetric synthesis route for synthesis of (S)-ketamine using a chiral reagent according to the strategy (Scheme 1), with good enantioselectivity (85% ee) and yield. In this procedure, the (S)-tert-butanesulfinamide (TBSA) acts as a chiral auxiliary reagent to generate (S)-ketamine. A series of new intermediates were synthesized and identified for the first time in this work (2–4). The monoketal intermediate (1) easily obtained after partial conversion of one ketone functional group of 1,2-cyclohexanedione into a ketal using ethylene glycol. The sulfinylimine (2) was obtained by condensation of (S)-tert-butanesulfinamide (TBSA) with (1), 4-dioxaspiro[4.5]decan-6-one in 90% yield. The (S)-N–tert-butanesulfinyl ketamine (3) was prepared on further reaction of sulfinylimine (2) with appropriate Grignard reagent (ArMgBr) in which generated chiral center in 85% yield and with 85% diastereoselectivity. Methylation of amine afforded the product (4). Finally, the sulfinyl- and ketal-protecting groups were removed from the compound (4) by brief treatment with stoichiometric quantities of HCl in a protic solvent gave the (S)-ketamine in near quantitative yield.
Esketamine was introduced for medical use in 1997.[1] In 2019, it was approved for use with other antidepressants, for the treatment of depression in adults in the United States.[9]
In August 2020, it was approved by the U.S. Food and Drug Administration (FDA) with the added indication for the short-term treatment of suicidal thoughts.[10]
In February 2019, an outside panel of experts recommended that the FDA approve the nasal spray version of esketamine,[13] provided that it be given in a clinical setting, with people remaining on site for at least two hours after. The reasoning for this requirement is that trial participants temporarily experienced sedation, visual disturbances, trouble speaking, confusion, numbness, and feelings of dizziness during immediately after.[14]
In January 2020, esketamine was rejected by the National Health Service of Great Britain. NHS questioned the benefits and claimed that it was too expensive. People who have been already using the medication were allowed to complete treatment if their doctors consider this necessary.[15]
Side effects
Most common side effects when used in those with treatment resistant depression include dissociation, dizziness, nausea, sleepiness, anxiety, and increased blood pressure.[16]
Pharmacology
Esketamine is approximately twice as potent as an anesthetic as racemic ketamine.[17] It is eliminated from the human body more quickly than arketamine (R(–)-ketamine) or racemic ketamine, although arketamine slows its elimination.[18]
A number of studies have suggested that esketamine has a more medically useful pharmacological action than arketamine or racemic ketamine[citation needed] but, in mice, that the rapid antidepressant effect of arketamine was greater and lasted longer than that of esketamine.[19] The usefulness of arketamine over eskatamine has been supported by other researchers.[20][21][22]
Esketamine inhibits dopamine transporters eight times more than arketamine.[23] This increases dopamine activity in the brain. At doses causing the same intensity of effects, esketamine is generally considered to be more pleasant by patients.[24][25] Patients also generally recover mental function more quickly after being treated with pure esketamine, which may be a result of the fact that it is cleared from their system more quickly.[17][26] This is however in contradiction with R-ketamine being devoid of psychotomimetic side effects.[27]
Unlike arketamine, esketamine does not bind significantly to sigma receptors. Esketamine increases glucose metabolism in frontal cortex, while arketamine decreases glucose metabolism in the brain. This difference may be responsible for the fact that esketamine generally has a more dissociative or hallucinogenic effect while arketamine is reportedly more relaxing.[26] However, another study found no difference between racemic and (S)-ketamine on the patient’s level of vigilance.[24] Interpretation of this finding is complicated by the fact that racemic ketamine is 50% (S)-ketamine.
History
Esketamine was introduced for medical use as an anesthetic in Germany in 1997, and was subsequently marketed in other countries.[1][28] In addition to its anesthetic effects, the medication showed properties of being a rapid-acting antidepressant, and was subsequently investigated for use as such.[8][12] In November 2017, it completed phase IIIclinical trials for treatment-resistant depression in the United States.[8][12]Johnson & Johnson filed a Food and Drug Administration (FDA) New Drug Application (NDA) for approval on September 4, 2018;[29] the application was endorsed by an FDA advisory panel on February 12, 2019, and on March 5, 2019, the FDA approved esketamine, in conjunction with an oral antidepressant, for the treatment of depression in adults.[9]
In the 1980s and ’90s, closely associated ketamine was used as a club drug known as “Special K” for its trip-inducing side effects.[30][31]
Society and culture
Names
Esketamine is the generic name of the drug and its INN and BAN, while esketamine hydrochloride is its BANM.[28] It is also known as S(+)-ketamine, (S)-ketamine, or (–)-ketamine, as well as by its developmental code name JNJ-54135419.[28][12]
Esketamine is marketed under the brand name Spravato for use as an antidepressant and the brand names Ketanest, Ketanest S, Ketanest-S, Keta-S for use as an anesthetic (veterinary), among others.[28]
Availability
Esketamine is marketed as an antidepressant in the United States;[9] and as an anesthetic in the European Union.[28]
^ Jump up to:abcdefghij Himmelseher S, Pfenninger E (December 1998). “[The clinical use of S-(+)-ketamine–a determination of its place]”. Anasthesiologie, Intensivmedizin, Notfallmedizin, Schmerztherapie. 33 (12): 764–70. doi:10.1055/s-2007-994851. PMID9893910.
^ Psychopharmacologic Drugs Advisory Committee (PDAC) and Drug Safety and Risk Management (DSaRM) Advisory Committee (February 12, 2019). “FDA Briefing Document” (PDF). Food and Drug Administration. Retrieved February 12, 2019. Meeting, February 12, 2019. Agenda Topic: The committees will discuss the efficacy, safety, and risk-benefit profile of New Drug Application (NDA) 211243, esketamine 28 mg single-use nasal spray device, submitted by Janssen Pharmaceutica, for the treatment of treatment-resistant depression.
^ Jump up to:ab Himmelseher S, Pfenninger E (December 1998). “[The clinical use of S-(+)-ketamine–a determination of its place]”. Anasthesiologie, Intensivmedizin, Notfallmedizin, Schmerztherapie (in German). 33 (12): 764–70. doi:10.1055/s-2007-994851. PMID9893910.
^ Ihmsen H, Geisslinger G, Schüttler J (November 2001). “Stereoselective pharmacokinetics of ketamine: R(–)-ketamine inhibits the elimination of S(+)-ketamine”. Clinical Pharmacology and Therapeutics. 70 (5): 431–8. doi:10.1067/mcp.2001.119722. PMID11719729.
^ Zhang JC, Li SX, Hashimoto K (January 2014). “R (-)-ketamine shows greater potency and longer lasting antidepressant effects than S (+)-ketamine”. Pharmacology, Biochemistry, and Behavior. 116: 137–41. doi:10.1016/j.pbb.2013.11.033. PMID24316345. S2CID140205448.
^ Jump up to:ab Doenicke A, Kugler J, Mayer M, Angster R, Hoffmann P (October 1992). “[Ketamine racemate or S-(+)-ketamine and midazolam. The effect on vigilance, efficacy and subjective findings]”. Der Anaesthesist (in German). 41 (10): 610–8. PMID1443509.
^ Pfenninger E, Baier C, Claus S, Hege G (November 1994). “[Psychometric changes as well as analgesic action and cardiovascular adverse effects of ketamine racemate versus s-(+)-ketamine in subanesthetic doses]”. Der Anaesthesist (in German). 43 Suppl 2: S68-75. PMID7840417.
^ Jump up to:ab Vollenweider FX, Leenders KL, Oye I, Hell D, Angst J (February 1997). “Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET)”. European Neuropsychopharmacology. 7 (1): 25–38. doi:10.1016/s0924-977x(96)00042-9. PMID9088882. S2CID26861697.
Mechanism of ActionOrnithine decarboxylase stimulants; Phosphokinase stimulants; Protein tyrosine kinase stimulants
Phase IIType 1 diabetes mellitus; Type 2 diabetes mellitus
28 Jul 2018No recent reports of development identified for phase-I development in Type-1 diabetes mellitus in Germany (SC, Injection)
28 Jul 2018No recent reports of development identified for phase-I development in Type-2-diabetes-mellitus in Denmark (SC, Injection)
11 Sep 2017Efficacy and adverse events data from a phase II trial in Type-2 diabetes mellitus presented at the 53rd Annual Meeting of the European Association for the Study of Diabetes (EASD-2017)
OI-338GT is a long-acting oral basal insulin analogue which had been in phase II clinical trials at Novo Nordisk for the treatment of patients with type 2 and type 1 diabetes. In 2016, the company discontinued the development of the product as the emergent product profile and required overall investments were not commercially viable in the increasingly challenging payer environment.
Recently, the first basal oral insulin (OI338) was shown to provide similar treatment outcomes to insulin glargine in a phase 2a clinical trial. Here, we report the engineering of a novel class of basal oral insulin analogues of which OI338, 10, in this publication, was successfully tested in the phase 2a clinical trial. We found that the introduction of two insulin substitutions, A14E and B25H, was needed to provide increased stability toward proteolysis. Ultralong pharmacokinetic profiles were obtained by attaching an albumin-binding side chain derived from octadecanedioic (C18) or icosanedioic acid (C20) to the lysine in position B29. Crucial for obtaining the ultralong PK profile was also a significant reduction of insulin receptor affinity. Oral bioavailability in dogs indicated that C18-based analogues were superior to C20-based analogues. These studies led to the identification of the two clinical candidates OI338 and OI320 (10 and 24, respectively).
Oral insulin 338 (I338) is a long-acting, basal insulin analogue formulated in a tablet with the absorption-enhancer sodium caprate. We investigated the efficacy and safety of I338 versus subcutaneous insulin glargine (IGlar) in patients with type 2 diabetes. METHODS: This was a phase 2, 8-week, randomised, double-blind, double-dummy, active-controlled, parallel trial completed at two research institutes in Germany. Insulin-naive adult patients with type 2 diabetes, inadequately controlled on metformin monotherapy or combined with other oral antidiabetic drugs (HbA1c 7·0-10·0%; BMI 25·0-40·0 kg/m(2)), were randomly assigned (1:1) to receive once-daily I338 plus subcutaneous placebo (I338 group) or once-daily IGlar plus oral placebo (IGlar group). Randomisation occurred by interactive web response system stratified by baseline treatment with oral antidiabetic drugs. Patients and investigators were masked to treatment assignment. Weekly insulin dose titration aimed to achieve a self-measured fasting plasma glucose (FPG) concentration of 4·4-7·0 mmol/L. The recommended daily starting doses were 2700 nmol I338 or 10 U IGlar, and maximum allowed doses throughout the trial were 16 200 nmol I338 or 60 U IGlar. The primary endpoint was treatment difference in FPG concentration at 8 weeks for all randomly assigned patients receiving at least one dose of trial product (ie, the full analysis set). The trial has been completed and is registered at ClinicalTrials.gov, number NCT02470039. FINDINGS: Between June 1, 2015, and Oct 19, 2015, 82 patients were screened for eligibility and 50 patients were randomly assigned to the I338 group (n=25) or the IGlar group (n=25). Mean FPG concentration at baseline was 9·7 (SD 2·8) in the I338 group and 9·1 (1·7) in the IGlar group. Least square mean FPG concentration at 8 weeks was 7·1 mmol/L (95% CI 6·4-7·8) in the I338 group and 6·8 mmol/L (6·5-7·1) in the IGlar group, with no significant treatment difference (0·3 mmol/L [-0·5 to 1·1]; p=0·46). I338 and IGlar were well tolerated by patients. Adverse events were reported in 15 (60%) patients in the I338 group and 17 (68%) patients in the IGlar group. The most common adverse events were diarrhoea (three [12%] patients in each group) and nasopharyngitis (five [20%] in the I338 group and two [8%] in the IGlar group). Most adverse events were graded mild (47 of 68 events), and no severe adverse events were reported. One patient in the IGlar group had a treatment-emergent serious adverse event (urogenital haemorrhage of moderate intensity, assessed by the investigator as unlikely to be related to treatment; the patient recovered). Incidence of hypoglycaemia was low in both groups (n=7 events in the I338 group; n=11 in the IGlar group), with no severe episodes. INTERPRETATION: I338 can safely improve glycaemic control in insulin-naive patients with type 2 diabetes with no evidence of a difference compared with insulin glargine, a widely used subcutaneously administered basal insulin. Further development of this particular oral insulin project was discontinued because I338 doses were high and, therefore, production of the required quantities of I338 for wide public use was deemed not commercially viable. Improvement of technologies involved in the product’s development is the focus of ongoing research. FUNDING: Novo Nordisk…..Halberg, I. B.; Lyby, K.; Wassermann, K.; Heise, T.; Zijlstra, E.; Plum-Mörschel, L. Efficacy and safety of oral basal insulin versus subcutaneous insulin glargine in type 2 diabetes: a randomised, double-blind, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 179– 188, DOI: 10.1016/s2213-8587(18)30372-3
ral insulin 338 is a novel tablet formulation of a long-acting basal insulin. This randomised, open-label, four-period crossover trial investigated the effect of timing of food intake on the single-dose pharmacokinetic properties of oral insulin 338. Methods: After an overnight fast, 44 healthy males received single fixed doses of oral insulin 338 administered 0, 30, 60 or 360 min before consuming a standardised meal (500 kcal, 57 energy percent [E%] carbohydrate, 13 E% fat, 30 E% protein). Blood samples for pharmacokinetic assessment were taken up to 288 h post-dose. Results: Total exposure (area under the concn.-time curve from time zero to infinity [AUCIns338,0-∞]) and max. concn. (Cmax,Ins338) of insulin 338 were both significantly lower for 0 vs. 360 min post-dose fasting (ratio [95% confidence interval (CI)]: 0.36 [0.26-0.49], p < 0.001, and 0.35 [0.25-0.49], p < 0.001, resp.). There were no significant differences in AUCIns338,0-∞ and Cmax,Ins338 for 30 or 60 vs. 360 min post-dose fasting (ratio [95% CI] 30 vs. 360 min: 0.85 [0.61-1.21], p = 0.36, and 0.86 [0.59-1.26], p = 0.42; ratio [95% CI] 60 vs. 360 min: 0.96 [0.72-1.28], p = 0.77, and 0.99 [0.75-1.31], p = 0.95). The mean half-life was ∼ 55 h independent of the post-dose fasting period. Oral insulin 338 was well-tolerated with no safety issues identified during the trial. Conclusions: Oral insulin 338 pharmacokinetics are not affected by food intake from 30 min after dosing, implying that patients with diabetes mellitus do not need to wait more than 30 min after a morning dose of oral insulin 338 before having their breakfast. This is considered important for convenience and treatment compliance. ClinicalTrials.gov identifier: NCT02304627./……Halberg, I. B.; Lyby, K.; Wassermann, K.; Heise, T.; Plum-Mörschel, L.; Zijlstra, E. The effect of food intake on the pharmacokinetics of oral basal insulin: A randomised crossover trial in healthy male subjects. Clin. Pharmacokinet. 2019, 58, 1497– 1504, DOI: 10.1007/s40262-019-00772-2
(E)-5-Methoxy-1-[4-(trifluoromethyl)phenyl]-1-pentanone O-(2-Aminoethyl)oxime1-Pentanone, 5-methoxy-1-[4-(trifluoromethyl)phenyl]-, O-(2-aminoethyl)oxime, (1E)-2-[({(1E)-5-Methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene}amino)oxy]ethanamine 2-{[(E)-{5-Methoxy-1-[4-(trifluoromethyl)phenyl]pentylidene}amino]oxy}ethanamine1-Pentanone, 5-methoxy-1-(4-(trifluoromethyl)phenyl)-, O-(2-aminoethyl)oxime, (E)- 387954739-18-3[RN]5583954[Beilstein]5-Methoxy-4′-(trifluoromethyl)valerophenone (E)-O-(2-aminoethyl)oximeA selective serotonin reuptake inhibitor that is used in the treatment of DEPRESSION and a variety of ANXIETY DISORDERS.
AbbVie Inc; Abbott Laboratories; Meiji Seika Pharma Co Ltd; Solvay SA
Launched (Obsessive compulsive disorder – EU – Dec-1983)
In the EU, the product is indicated for the treatment of obsessive compulsive disorder (OCD) and for the treatment of major depressive disorder (MDD)
In Japan, Luvox is indicated for the treatment of adult or pediatric OCD, social anxiety disorder (SAD) and MDD
USFDA The drug was approved for the treatment of OCD and SAD in April 2008
CHINA
In 2000, the drug was launched in China for the treatment of OCD and MDD
Patents and Generics
FDA exclusivity expired in the US in June 2000. Generic versions have been on the market since that time. Generic fluvoxamine was still available in the US by May 2007, despite the fact the Solvay/Jazz product had not been relaunched . By October 2004, the drug was also off patent in most European countries .
Medical uses
Fluvoxamine is approved in the United States for OCD,[9][6] and social anxiety disorder.[10] In other countries (e.g., Australia,[11][12] the UK,[13] and Russia[14]) it also has indications for major depressive disorder. In Japan it is currently[when?] approved to treat OCD, SAD and MDD.[15][16] Fluvoxamine is indicated for children and adolescents with OCD.[17] The drug works long-term, and retains its therapeutic efficacy for at least one year.[18] It has also been found to possess some analgesic properties in line with other SSRIs and tricyclic antidepressants.[19][20][21]
There is tentative evidence that fluvoxamine is effective for social phobia in adults.[22] Fluvoxamine is also effective for GAD, SAD, panic disorder and separation anxiety disorder in children and adolescents.[23] There is tentative evidence that fluvoxamine may help some people with negative symptoms of chronic schizophrenia.[24][25]
A double-blind controlled study found that fluvoxamine may prevent clinical deterioration in outpatients with symptomatic COVID-19. The study had important limitations: it was run fully remotely; it had a small sample size (150) and short follow-up duration (15 days).[26] The accompanying editorial noted that, although this study is important enough to choose out of more than 10,000 other COVID-19 related submissions, it “presents only preliminary information” and “the findings should be interpreted as only hypothesis generating; they should not be used as the basis for current treatment decisions.”[27] Similarly, the study authors themselves cautioned that “the trial’s results should not be treated as a measure of fluvoxamine’s effectiveness against COVID-19 but as an encouraging indicator that the drug warrants further testing.”[28] A prospective open-labelled cohort study showed similar results.[29]
Adverse effects
Gastrointestinal side effects are more common in those receiving fluvoxamine than with other SSRIs.[30] Otherwise, fluvoxamine’s side-effect profile is very similar to other SSRIs.[2][9][11][13][31][32]Common (1–10% incidence) adverse effects
Fluvoxamine and ramelteon coadministration is not indicated.[52][53]
Fluvoxamine has been observed to increase serum concentrations of mirtazapine, which is mainly metabolized by CYP1A2, CYP2D6, and CYP3A4, by 3- to 4-fold in humans.[54] Caution and adjustment of dosage as necessary are warranted when combining fluvoxamine and mirtazapine.[54]
Fluvoxamine seriously affects the pharmacokinetics of tizanidine and increases the intensity and duration of its effects. Because of the potentially hazardous consequences, the concomitant use of tizanidine with fluvoxamine, or other potent inhibitors of CYP1A2, should be avoided.[55]
Fluvoxamine was developed by Kali-Duphar,[63] part of Solvay Pharmaceuticals, Belgium, now Abbott Laboratories, and introduced as Floxyfral in Switzerland in 1983.[63] It was approved by the U.S. Food and Drug Administration (FDA) in 1994, and introduced as Luvox in the US.[64] In India, it is available, among several other brands, as Uvox by Abbott.[65] It was one of the first SSRI antidepressants to be launched, and is prescribed in many countries to patients with major depression.[66] It was the first SSRI, a non-TCA drug, approved by the U.S. FDA specifically for the treatment of OCD.[67] At the end of 1995, more than ten million patients worldwide had been treated with fluvoxamine.[68][failed verification] Fluvoxamine was the first SSRI to be registered for the treatment of obsessive compulsive disorder in children by the FDA in 1997.[69] In Japan, fluvoxamine was the first SSRI to be approved for the treatment of depression in 1999[70][71] and was later in 2005 the first drug to be approved for the treatment of social anxiety disorder.[72] Fluvoxamine was the first SSRI approved for clinical use in the United Kingdom.[73]
Society and culture
Manufacturers include BayPharma, Synthon, and Teva, among others.[74]
SYN
SYN
J. Zhejiang Univ. (Medical Sci.) (2003), 32 (5), 441-442
The present invention relates to an improved and industrially applicable process for the preparation of fluvoxamine maleate of formula I,
Fluvoxamine or (E)-5-methoxy-1 -[4-(trifluoromethyl)phenyl]pentan- 1 -one-O-2-aminoethyl oxime is an antidepressant which functions as a selective serotonin reuptake inhibitor (SSRI). Fluvoxamine is used for the treatment of major depressive disorder (MDD), obsessive compulsive disorder (OCD), and anxiety disorders such as panic disorder and post-traumatic stress disorder (PTSD). Fluvoxamine CR (controlled release) is approved to treat social anxiety disorder.
Fluvoxamine maleate and compounds were first disclosed in US patent 4,085,225. According to said patent, Fluvoxamine maleate prepared by alkylation reaction of 5-methoxy-4′-trifluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in dimethylformamide in the presence of a base such as potassium hydroxide powder for two days at 25°C.
Subsequently the solvent is removed under vacuum then the residue is acidified and extracted with ether to remove the unreacted oxime followed by basification. The obtained fluvoxamine base in ether extract is washed with sodium bicarbonate solution. The fluvoxamine base is then treated with maleic acid in absolute ethanbl and the residue obtained by concentration under vacuum is recrystallized from acetonitrile to obtain fluvoxamine maleate. The process is very much tedious, time consuming as it requires two days for the reaction completion. Operations like removal of dimethylformamide, ether, ethanol makes process cumbersome at plant level. Requirement of
various solvents lead the process to be non-eco-friendly. Moreover the patent is silent about yield and purity of the product.
In an alternate route described in US patent 4,085,225, the oxime of formula III is converted to formula I in a five step process i.e. alkylation of formula III with ethylene oxide. The reaction solvent is ethanol in which lithium is already dissolved. The reaction further involves addition of acetic acid to give the hydroxyethyl compound of formula A as oil. The compound of formula A is purified chromatographically over the silica gel, which is converted to a mesylate compound of formula B by treating with methanesulfonyl chloride and triethylamine at -5 to 0°C, then aminated with ammonia in methanol at 100°C using autoclave for 16 hours followed by removal of methanol and extraction in ether to give fluvoxamine base.
The base is then converted to the maleate salt formula I, which is finally purified by recrystallization from acetonitrile.
There are lots of disadvantages involve like more unit operations, use of various solvents and handling of ethylene oxide which is also known for its carcinogen effect. More unit operations lead to long occupancy of reactors in the plant as well as man power, high energy consumption and require bigger plant. These all parameters make the process commercially unviable as wel l as environmentally non-feasible. Further, purification of the compound of formula A requires cumbersome technique i.e chromatography over silica gel as well as lengthy work-up procedure in U.S. Pat. No. 4,085,225 requires complete removal of organic solvents at various stages.
US patent 6,433,225 discloses the process for preparing fluvoxamine maleate, prepared by alkylating 5-methoxy-4′-trtfluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in toluene and PEG-400 (polyethyleneglycol-400) as facilitator in the presence of a base potassium hydroxide powder at 30-35°C to obtain fluvoxamine base in
toluene layer is then treated with maleic acid in water. The precipitated fluvoxamine maleate is filtered and washed with toluene and dried. The obtained dried cake recrystallized with water to get fluvoxamine maleate. The process disclosed in the patent is silent about actual purity of the product. As per our scientist’s observation alkylation reaction at the temperature of 30-35°C may lead to non completion of reaction and results lower yield. Additional step of purification may further lead to loss of yield.
Thus, present invention fulfills the need of the art and provides an improved and industrially applicable process for preparation of fluvoxamine maleate, which provides fluvoxamine maleate in high purity and overall good yield.
EXAMPLES:
Stage – 1 : Preparation of (1E)-N-hydroxy-5-methoxy-1-(4-trifluoromethyI pheny 1) pentan-1-imine formula III
To a stirred solution of 5-methoxy- 1 -(4-trifluoromethylphenyl) pentan-1 -one ( 150 gm) in methanol (750 ml), sodium carbonate (granule) (72 gm) and hydroxylamine hydrochloride (59.64 gm) were added at temperature 25-30°C. The reaction mass was heated 45-50°C for 10- 15 minutes followed by maintaining the reaction mass at temperature 45-50°C for 8-9 hours under stirring. The reaction mass was cooled to 25-30°C and filtered under vacuum to remove unreacted inorganic matter, then distilled out the methanol completely from the collected filtrate under vacuum at temperature below 50°C. The obtained slurry was cooled to 25-30°C and water (300 ml) was added into the residue followed by the addition of hexane (300×2 ml) and stirred for 30 minutes. The layers were separated. The collected organic layer was stirred for 5- 10 minutes at temperature 25-30°C followed by cooling the mass at temperature -5°C to – 10°C, stirred for 30-40 minutes and filtered at the same temperature. The product was suck dried at -5 to -10°C and further in vacuum at 25-30°C for 2-3 hours to give 138 – 142 gm of title compound. HPLC purity: >98.5%
Stage – 2: Preparation of crude fluvoxamine maleate formula I
To a prepared solution of dimethyl sulphoxide (575 ml), potassium hydroxide flakes ( 1 14.64 gm) and water (69 ml), stage-1 (1 15 gm) was added at temperature 40-45°C. The reaction mixture was stirred to get clear solution followed by adding 2-chloroethylamine hydrochloride (86.36 gm) drop wise into the reaction mixture at temperature 40-45°C and maintained for 1 -2 hour. Water (1 150 ml) was added in to the reaction mixture at temperature 25-30°C and stirred for 20-25 minutes. Then toluene (575 ml x 2) was added and stirred for 30 minutes and preceded for separation of layers followed by washing the toluene layer with water ( 1 1 50 x 5 ml). The solution of maleic acid (48.47 gm) dissolved in water (98 ml) was added into above obtained toluene layer and stirred at temperature 25-30°C for 2-3 hours. The reaction mixture was cooled to 0-5°C and maintained for 30-40 minutes at the same temperature. The obtained material was washed with toluene, filtered and suck dried. The wet cake was then added hexane (600 ml) and stirred for 30 minutes at temperature 25-30°C, filtered, washed with hexane and dried to get 161 gm of title compound. HPLC purity: >98.5%
Stage – 3: Preparation of pure fluvoxamine maleate formula I
In to the reaction assembly, water (600 ml) was added and heated to 40-45°C. Stage -2 ( 1 50 gm) was added into the hot water under stirring. The reaction mixture was stirred for 5- 10 minutes, filtered and cooled to 25°C. Toluene (68 ml) was added into the reaction mixture at temperature 25°C and stirred for 30 minutes. Filtered the solid, washed with 10-15°C chilled water and dried to get the pure 127.5 gm fluvoxamine maleate. HPLC purity: >99.8%
Process for isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one formula II
To a solution of cone. HCl (600 ml) and water ( 160 ml), organic residue (250 gm) of ( 1 £)+( 1 Z) of 1 -N-hydroxy-5-methoxy- 1 -[4-(trifluoromethyl) phenyl]pentan-1 -imine and traces of 5-methoxy- 1 -[4-(trifluoromethyl)phenyl]pentan- 1-one (obtained after hexane recovery from stage-1 filtrate) was added at temperature 25-30°C under stirring. The reaction mixture was heated to 67-75°C and maintained for 13-14 hours followed by cool ing the reaction mixture at temperature 25-30°C. Then after hexane (500 x 2 ml) was added into the reaction mixture and stirred for 15 minutes at 25-30°C. The organic layers were separated and sodium bicarbonate solution (25 gm sodium bicarbonate dissolved in 250 ml water) was added into the hexane layer and stirred for 15 minutes. The layers were separated and water (250ml) was added into hexane layer and stirred for 15 minutes at temperature 25-30°C. Further the layers were separated and hexane layer was added activated charcoal ( 12.5 gm) and stirred for 20-30 minutes at temperature 30-35°C. The reaction mixture was filtered and stirred for 5-10 minutes at 25-30°C followed by cooling at 0 to -5°C and stirred for 30-40 minutes at 0 to -5°C. The reaction mixture was filtered and dried to get 150 to l 75 gm of title compound. HPLC purity: >99%.
Fluvoxamine or (E)-5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one-O-2-aminoethyl oxime is an antidepressant which functions as a selective serotonin reuptake inhibitor (SSRI). Fluvoxamine is used for the treatment of major depressive disorder (MDD), obsessive compulsive disorder (OCD), and anxiety disorders such as panic disorder and post-traumatic stress disorder (PTSD). Fluvoxamine CR (controlled release) is approved to treat social anxiety disorder.
Fluvoxamine maleate and compounds were first disclosed in U.S. Pat. No. 4,085,225. According to said patent, Fluvoxamine maleate prepared by alkylation reaction of 5-methoxy-4′-trifluoromethylvalerophenone oxime, compound of formula III with 2-chloroethylamine hydrochloride in dimethylformamide in the presence of a base such as potassium hydroxide powder for two days at 25° C.
Subsequently the solvent is removed under vacuum then the residue is acidified and extracted with ether to remove the unreacted oxime followed by basification. The obtained fluvoxamine base in ether extract is washed with sodium bicarbonate solution. The fluvoxamine base is then treated with maleic acid in absolute ethanol and the residue obtained by concentration under vacuum is recrystallized from acetonitrile to obtain fluvoxamine maleate. The process is very much tedious, time consuming as it requires two days for the reaction completion. Operations like removal of dimethylformamide, ether, ethanol makes process cumbersome at plant level. Requirement of various solvents lead the process to be non-eco-friendly. Moreover the patent is silent about yield and purity of the product.
In an alternate route described in U.S. Pat. No. 4,085,225, the mine of formula III is converted to formula I in a five step process i.e. alkylation of formula III with ethylene oxide. The reaction solvent is ethanol in which lithium is already dissolved. The reaction further involves addition of acetic acid to give the hydroxyethyl compound of formula A as oil. The compound of formula A is purified chromatographically over the silica gel, which is converted to a mesylate compound of formula B by treating with methanesulfonyl chloride and triethylamine at −5 to 0° C., then aminated with ammonia in methanol at 100° C. using autoclave for 16 hours followed by removal of methanol and extraction in ether to give fluvoxamine base.
The base is then converted to the maleate salt formula I, which is finally purified by recrystallization from acetonitrile.
There are lots of disadvantages in like more unit operations, use of various solvents and handling of ethylene oxide which is also known for its carcinogen effect. More unit operations lead to long occupancy of reactors in the plant as well as man power, high energy consumption and require bigger plant. These all parameters make the process commercially unviable as well as environmentally non-feasible. Further, purification of the compound of formula A requires cumbersome technique i.e chromatography over silica gel as well as lengthy work-up procedure in U.S. Pat. No. 4,085,225 requires complete removal of organic solvents at various stages.
U.S. Pat. No. 6,433,225 discloses the process for preparing fluvoxamine maleate, prepared by alkylating 5-methoxy-4′-trifluoromethylvalerophenone oxime compound of formula III with 2-chloroethylamine hydrochloride in toluene and PEG-400 (polyethyleneglycol-400) as facilitator in the presence of a base potassium hydroxide powder at 30-35°C. to obtain fluvoxamine base in toluene layer is then treated with maleic acid in water. The precipitated fluvoxamine maleate is filtered and washed with toluene and dried. The obtained dried cake recrystallized with water to get fluvoxamine maleate. The process disclosed in the patent is silent about actual purity of the product. As per our scientist’s observation alkylation reaction at the temperature of 30-35° C. may lead to non completion of reaction and results lower yield. Additional step of purification may further lead to loss of yield.
EXAMPLES
Stage-1: Preparation of (1 E)-N-hydroxy-5-methoxy-1-(4-trifluoromethyl phenyl)pentan-1-imine Formula III
To a stirred solution of 5-methoxy-1-(4-trifluoromethylphenyl)pentan-1one (150 gm) in methanol (750 ml), sodium carbonate (granule) (72 gm) and hydroxylamine hydrochloride (59.64 gm) were added at temperature 25-30° C. The reaction mass was heated 45-50° C. for 10-15 minutes followed by maintaining the reaction mass at temperature 45-50° C. for 8-9 hours under stirring. The reaction mass was cooled to 25-30° C. and filtered under vacuum to remove unreacted inorganic matter, then distilled out the methanol completely from the collected filtrate under vacuum at temperature below 50° C. The obtained slurry was cooled to 25-30° C. and water (300 ml) was added into the residue followed by the addition of hexane (300×2 ml) and stirred for 30 minutes. The layers were separated. The collected organic layer was stirred for 5-10 minutes at temperature 25-30° C. followed by cooling the mass at temperature −5° C. to −10° C., stirred for 30-40 minutes and filtered at the same temperature. The product was suck dried at −5 to −10° C. and further in vacuum at 25-30° C. for 2-3 hours to give 138-142 gm of title compound. HPLC purity: >98.5%
Stage-2: Preparation of Crude Fluvoxamine Maleate Formula I
To a prepared solution of dimethyl sulphoxide (575 ml), potassium hydroxide flakes (114.64 gm) and water (69 ml), stage-1 (115 gm) was added at temperature 40-45° C. The reaction mixture was stirred to get clear solution followed by adding 2-chloroethylamine hydrochloride (8636 gm) drop wise into the reaction mixture at temperature 40-45° C. and maintained for 1-2 hour. Water (1150 ml) was added in to the reaction mixture at temperature 25-30° C. and stirred for 20-25 minutes. Then toluene (575 ml×2) was added and stirred for 30 minutes and preceded for separation of layers followed by washing the toluene layer with water (1150×5 ml). The solution of maleic acid (48.47 gm) dissolved in water (98 ml) was added into above obtained toluene layer and stirred at temperature 25-30° C. for 2-3 hours. The reaction mixture was cooled to 0-5° C. and maintained for 30-40 minutes at the same temperature. The obtained material was washed with toluene, filtered and such dried. The wet cake was then added hexane (600 ml) and stirred for 30 minutes at temperature 25-30° C., filtered, washed with hexane and dried to get 161 gm of title compound. HPLC purity: >98.5%
Stage-3: Preparation of Pure Fluvoxamine Maleate Formula I
In to the reaction assembly, water (600 ml) was added and heated to 40-45° C. Stage-2 (150 gm) was added into the hot water under stirring. The reaction mixture was stirred for 5-10 minutes, filtered and cooled to 25° C. Toluene (68 ml) was added into the reaction mixture at temperature 25° C. and stirred for 30 minutes. Filtered the solid, washed with 10-15° C. chilled water and dried to get the pure 127.5 gm fluvoxamine maleate. HPLC purity: >99.8%
Process for isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one Formula II
To a solution of conc. HCl (600 ml) and water (160 organic residue (250 gm) of (1 E)+(1 Z) of 1-N-hydroxy-5-methoxy-1-[4trifluoromethyl)phenyl]pentan-1-imine and traces of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one (obtained after hexane recovery from stage-1 filtrate) was added at temperature 25-30° C. under stirring. The reaction mixture was heated to 67-75° C. and maintained for 13-14 hours followed by cooling the reaction mixture at temperature 25-30° C. Then after hexane (500×2 ml) was added into the reaction mixture and stirred for 15 minutes at 25-30° C. The organic layers were separated and sodium bicarbonate solution (25 gm sodium bicarbonate dissolved in 250 ml water) was added into the hexane layer and stirred for 15 minutes. The layers were separated and water (250 ml) was added into hexane layer and stirred for 15 minutes at temperature 25-30° C. Further the layers were separated and hexane layer was added activated charcoal (12.5 gm) and stirred for 20-30 minutes at temperature 30-35° C. The reaction mixture was filtered and stirred for 5-10 minutes at 25-30° C. followed by cooling at 0 to −5° C. and stirred for 30-40 minutes at 0 to −5° C. The reaction mixture was filtered and dried to get 150 to 175 gm of title compound. HPLC purity: >99%. Claims (5)Hide Dependent
We claim:1. An improved process for the preparation of fluvoxamine maleate of formula I,
wherein the improvements comprises the steps of:a). condensing the compound of formula II,
with hydroxylamine hydrochloride in the presence of sodium carbonate granules at temperature 45-50° C. in suitable solvent to form a compound of formula III, wherein the compound of formula III comprises a mixture of (1E)+(1Z) isomers of 1-N-hydroxy-5-methoxy-1-[4(trifluoromethyl)phenyl]pentan-1-imine, and wherein the mixture of (1E)+(1Z) isomers of 1-N-hydroxy-5-methoxy-1-[4(trifluoromethyl)phenyl]pentan-1-imine comprises 98% of E-isomer and 2% of Z-isomer;
b). isolating compound of formula III;c). treating compound of formula III with 2-chloroethylamine hydrochloride in the presence of base in suitable solvent at 40-45° C. to form compound of formula IV;
d). extracting compound of formula IV with suitable solvent to form an organic layer;e). treating organic layer of step d) with maleic acid;f). isolating crude fluvoxamine maleate of formula I; andg). optionally purifying fluvoxamine maleate of formula I.
2. The process according to claim 1, wherein in step a), said suitable solvent is selected from the group consisting of alcohol, ketone, nitrile, and hydrocarbons in any suitable proportion or mixtures thereof;in step c), said base is selected from the group consisting of sodium hydroxide, potassium hydroxide, lithium hydroxide, sodium carbonate, potassium carbonate, lithium carbonate, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate, triethylamine and diisopropylethyamine;in step c), said solvent is selected from the group consisting of dimethylformamide (DMF), dimethylsulphoxide (DMSO) and hexamethylphosphoramide (HMPA) in any suitable proportion or mixtures thereof; andin step d) said suitable solvent is selected from the group consisting of toluene and xylene.3. A process for the isolation of 5-methoxy-1-[4-(trifluoromethyl)phenyl]pentan-1-one of formula II from mixture of (1E)+(1Z) of 1-N -hydroxy-5-methoxy-1-[4-(trifluoromethyl) phenyl]pentan-1-imine of formula III by treating compound of formula III with aqueous hydrochloric acid, wherein the mixture of (1E)+(1Z) of 1-N-hydroxy-5-methoxy-1-[4-(trifluoromethyl) phenyl]pentan-1-imine of formula III comprises 98% of E-isomer and 2% of Z-isomer.4. The process according to claim 3, wherein the reaction is performed at temperature 65-75°C.5. The process according to claim 1, wherein in step a), said suitable solvent is methanol. Publication numberPriority datePublication dateAssigneeTitleUS4081551A *1975-03-201978-03-28U.S. Philips CorporationOxime ethers having anti-depressive activityUS4085225A1975-03-201978-04-18U.S. Philips CorporationOxime ethers having anti-depressive activityCN1079733A *1993-04-081993-12-22中国科学院成都有机化学研究所The synthetic method of a-benzoin oximeUS6433225B11999-11-122002-08-13Sun Pharamaceutical Industries, Ltd.Process for the preparation of fluvoxazmine maleateCN101654419A *2009-09-122010-02-24西北师范大学Preparation method of fluvoxamine maleate Syn
To a stirred mixture of toluene (1.20 lit.), PEG-400 (0.4 lit) and powdered potassium hydroxide (86.0 g on 100% basis, 1.53 mol.) at ambient temperature is added 5-methoxy-4′-trifluoromethylvalerophenone oxime (100 g, 0.363 mol.), followed by 2-chloroethyl amine hydrochloride (50.56 g, 0.435 mol.). The mixture is stirred at 30-35° C. for 2 hours. Water (1.2 lit.) is then added, stirred for 30 mins. and the aqueous layer is separated out. The organic layer is washed with water (˜3×500 ml) until the washings are neutral. To the washed organic layer is added a solution of maleic acid (14.14 g, 0.363 mol.) in water (65 ml) and the mixture is stirred at 25-30° C. temperature for 2 hours, then cooled to 5-10° C. when the maleate salt crystallizes out. The crystallized fluvoxamine maleate is filtered, washed with toluene (200 ml) and sucked to dryness. The crude fluvoxamine maleate thus obtained is dissolved in water (300 ml) at 50-55° C. to get a clear solution, then gradually cooled to 5-8° C. and then further stirred at this temperature for 2 hours. The recrystallised fluvoxamine maleate is filtered, washed with chilled water (5° C., 100 ml) and sucked dry. The product is finally dried at 50-55° C. to constant weight. The fluvoxamine maleate obtained complies with the specifications of British Pharmacopoeia, 1999.EXAMPLE 2
This process when scaled up in pilot plant on 4.0 kg scale input of 5-methoxy-4′-trifluoromethylvalerophenone oxime gave 4.5 kg (71.2%) of fluvoxamine maleate, complying to the specifications of British Pharmacopoeia, 1999.
EXAMPLE 15-Methoxy-4′-trifluoromethylvalerophenone O-(2-aminoethyl) oxime maleate (1:1).
20.4 Mmol (5.3 g) of 5-methoxy-4′-trifluoromethylvalerophenone (melting point 43°-44° C), 20.5 mmol (3.1 g) of 2-aminooxyethylaminedihydrochloride and 10 ml of pyridine were refluxed for 15 hours in 20 ml of absolute ethanol. After evaporating the pyridine and the ethanol in vacuo, the residue was dissolved in water. This solution was washed with petroleum ether and 10 ml of 50% sodium hydroxide solution were then added. Then three extractions with 40 ml of ether were carried out. The ether extract was washed successively with 20 ml of 5% sodium bicarbonate solution and 20 ml of water. After drying on sodium sulphate, the ether layer was evaporated in vacuo. Toluene was then evaporated another three times (to remove the pyridine) and the oil thus obtained was dissolved in 15 ml of absolute ethanol. An equimolar quantity of maleic acid was added to said solution and the solution was then heated until a clear solution was obtained. The ethanol was then removed in vacuo and the residue was crystallized from 10 ml of acetonitrile at +5° C. After sucking off and washing with cold acetonitrile, it was dried in air. The melting point of the resulting title compound was 120°-121.5° C.
^ Figgitt DP, McClellan KJ (October 2000). “Fluvoxamine. An updated review of its use in the management of adults with anxiety disorders”. Drugs. 60 (4): 925–54. doi:10.2165/00003495-200060040-00006. PMID11085201.
^ Jump up to:ab Rossi S, ed. (2013). Australian Medicines Handbook (2013 ed.). Adelaide: The Australian Medicines Handbook Unit Trust. ISBN978-0-9805790-9-3.
^ “US-FDA Fluvoxamine Product Insert”. March 2005.
^ Wilde MI, Plosker GL, Benfield P (November 1993). “Fluvoxamine. An updated review of its pharmacology, and therapeutic use in depressive illness”. Drugs. 46(5): 895–924. doi:10.2165/00003495-199346050-00008. PMID7507038.
^ Kwasucki J, Stepień A, Maksymiuk G, Olbrych-Karpińska B (2002). “[Evaluation of analgesic action of fluvoxamine compared with efficacy of imipramine and tramadol for treatment of sciatica–open trial]”. Wiadomosci Lekarskie. 55 (1–2): 42–50. PMID12043315.
^ Schreiber S, Pick CG (August 2006). “From selective to highly selective SSRIs: a comparison of the antinociceptive properties of fluoxetine, fluvoxamine, citalopram and escitalopram”. European Neuropsychopharmacology. 16 (6): 464–8. doi:10.1016/j.euroneuro.2005.11.013. PMID16413173. S2CID39278756.
^ Coquoz D, Porchet HC, Dayer P (September 1993). “Central analgesic effects of desipramine, fluvoxamine, and moclobemide after single oral dosing: a study in healthy volunteers”. Clinical Pharmacology and Therapeutics. 54 (3): 339–44. doi:10.1038/clpt.1993.156. PMID8375130. S2CID8229797.
^ Brayfield A, ed. (13 August 2013). Fluoxetine Hydrochloride. Martindale: The Complete Drug Reference. London, UK: Pharmaceutical Press. Retrieved 24 November 2013.
^ Taylor D, Paton C, Shitij K (2012). The Maudsley prescribing guidelines in psychiatry. West Sussex: Wiley-Blackwell. ISBN978-0-470-97948-8.
^ Hemeryck A, Belpaire FM (February 2002). “Selective serotonin reuptake inhibitors and cytochrome P-450 mediated drug-drug interactions: an update”. Current Drug Metabolism. 3 (1): 13–37. doi:10.2174/1389200023338017. PMID11876575.
^ Suzuki Y, Shioiri T, Muratake T, Kawashima Y, Sato S, Hagiwara M, Inoue Y, Shimoda K, Someya T (April 2003). “Effects of concomitant fluvoxamine on the metabolism of alprazolam in Japanese psychiatric patients: interaction with CYP2C19 mutated alleles”. European Journal of Clinical Pharmacology. 58 (12): 829–33. doi:10.1007/s00228-003-0563-9. PMID12698310. S2CID32559753.
^ Fleishaker JC, Hulst LK (1994). “A pharmacokinetic and pharmacodynamic evaluation of the combined administration of alprazolam and fluvoxamine”. European Journal of Clinical Pharmacology. 46 (1): 35–9. doi:10.1007/bf00195913. PMID8005185. S2CID2161450.
^ Obach RS, Ryder TF (August 2010). “Metabolism of ramelteon in human liver microsomes and correlation with the effect of fluvoxamine on ramelteon pharmacokinetics”. Drug Metabolism and Disposition. 38 (8): 1381–91. doi:10.1124/dmd.110.034009. PMID20478852. S2CID8421997.
^ Jump up to:ab Anttila AK, Rasanen L, Leinonen EV (October 2001). “Fluvoxamine augmentation increases serum mirtazapine concentrations three- to fourfold”. The Annals of Pharmacotherapy. 35 (10): 1221–3. doi:10.1345/aph.1A014. PMID11675851. S2CID44807359.
^ Granfors MT, Backman JT, Neuvonen M, Ahonen J, Neuvonen PJ (April 2004). “Fluvoxamine drastically increases concentrations and effects of tizanidine: a potentially hazardous interaction”. Clinical Pharmacology and Therapeutics. 75(4): 331–41. doi:10.1016/j.clpt.2003.12.005. PMID15060511. S2CID25781307.
^ Ishikawa M, Ishiwata K, Ishii K, Kimura Y, Sakata M, Naganawa M, et al. (October 2007). “High occupancy of sigma-1 receptors in the human brain after single oral administration of fluvoxamine: a positron emission tomography study using [11C]SA4503”. Biological Psychiatry. 62 (8): 878–83. doi:10.1016/j.biopsych.2007.04.001. PMID17662961. S2CID728565.
^ Schatzberg AF, Nemeroff CB (2009). The American Psychiatric Publishing textbook of psychopharmacology (4th ed.). Arlington, VA: American Psychiatric Pub. p. 354. ISBN978-1-585-62386-0. OCLC320111564.
^ Jump up to:ab Hashimoto K (September 2009). “Sigma-1 receptors and selective serotonin reuptake inhibitors: clinical implications of their relationship”. Central Nervous System Agents in Medicinal Chemistry. 9 (3): 197–204. doi:10.2174/1871524910909030197. PMID20021354.
^ Hindmarch I, Hashimoto K (April 2010). “Cognition and depression: the effects of fluvoxamine, a sigma-1 receptor agonist, reconsidered”. Human Psychopharmacology. 25 (3): 193–200. doi:10.1002/hup.1106. PMID20373470. S2CID26491662.
EverolimusCAS Registry Number: 159351-69-6CAS Name: 42-O-(2-Hydroxyethyl)rapamycinAdditional Names: 40-O-(2-hydroxyethyl)rapamycinManufacturers’ Codes: RAD-001; SDZ RADTrademarks: Certican (Novartis)Molecular Formula: C53H83NO14Molecular Weight: 958.22Percent Composition: C 66.43%, H 8.73%, N 1.46%, O 23.38%Literature References: Macrolide immunosuppressant; derivative of rapamycin, q.v. Inhibits cytokine-mediated lymphocyte proliferation. Prepn: S. Cottens, R. Sedrani, WO9409010; eidem, US5665772 (1994, 1997 both to Sandoz). Pharmacology: W. Schuler et al., Transplantation64, 36 (1997). Whole blood determn by LC/MS: N. Brignol et al., Rapid Commun. Mass Spectrom.15, 898 (2001); by HPLC: S. Baldelli et al., J. Chromatogr. B816, 99 (2005). Clinical pharmacokinetics in combination with cyclosporine: J. M. Kovarik et al., Clin. Pharmacol. Ther.69, 48 (2001). Clinical study in prevention of cardiac-allograft vasculopathy: H. J. Eisen et al.,N. Engl. J. Med.349, 847 (2003). Review: F. J. Dumont et al., Curr. Opin. Invest. Drugs2, 1220-1234 (2001); B. Nashan, Ther. Drug Monit.24, 53-58 (2002).Therap-Cat: Immunosuppressant.Keywords: Immunosuppressant.эверолимус[Russian][INN]إيفيروليموس[Arabic][INN]依维莫司[Chinese][INN]Trade Name:Certican® / Zortress® / Afinitor®MOA:mTOR inhibitorIndication:Rejection of organ transplantation; Renal cell carcinoma; Advanced renal cell carcinoma (RCC); Advanced breast cancer; Pancreatic cancer; Renal angiomyolipoma; Tuberous sclerosis complex (TSC); Rejection in heart transplantation; Rejection of suppression renal transplantation; Subependymal giant cell astrocytoma; neuroendocrine tumors (NET); Advanced gastrointestinal tumorsStatus:ApprovedCompany:Novartis (Originator)Sales:$1,942 Million (Y2015); $1,902 Million (Y2014); $1,558 Million (Y2013); $1,007 Million (Y2012); $630 Million (Y2011);ATC Code:L04AA18Approved Countries or Area
Rejection of organ transplantation, Renal cell carcinoma
Tablet
0.25 mg/0.5 mg/0.75 mg
Novartis
clip
Active Substance The active substance Everolimus is a hydroxyethyl derivative of rapamycin, which is a macrolide, isolated from the micro-organism Streptomyces hygroscopicus. The guideline, impurities in new active substances ICHQ 3A (R), does not apply to active substance of fermented origin. Everolimus (INN) or 42-O-(2-hydroxyethyl)-rapamycin (chemical name) or C5 3H8 3N O1 4 has been fully described. The molecule is amorphous and is stabilised with an antioxidant. Its physico-chemical properties including parameters such as solubility, pH, specific rotation, potential polymorphism and potential isomerism have been fully characterised. Everolimus is a white to faintly yellow amorphous powder. It is almost insoluble in water, is unstable at temperatures above 25 °C and is sensitive to light. In addition, possible isomerism has been investigated. Everolimus contains 15 asymmetric carbon atoms and 4 substituted double bonds. The configuration of the asymmetric carbon atoms and the double bonds is guaranteed by the microbial origin of Rapamycin. The configuration is not affected by the chemical synthesis. Polymorphism has been comprehensively discussed and it was demonstrated that the molecule domain remains amorphous.
Synthesis of Everolimus The manufacturing process consists of four main steps, (1) fermentation, (2) extraction of rapamycin from the fermentation broth, (3) chemical modification of rapamycin starting material, (4) purification of crude everolimus and stabilisation with BHT. The choice of the stabilizer has been sufficiently explained and justified by experimental results. Interactions products of Everolimus and the antioxidant were not detected, or were below detection limit. Rapamycin, obtained by a fermentation process, was used as the starting material. Reaction conditions and the necessary in-process controls are described in detail. Adequate specifications for starting materials and isolated intermediates and descriptions of the test procedures have been submitted. Control of the quality of solvents, reagents and auxiliary materials used in the synthesis has been adequately documented. It is stated by the manufacturer of rapamycin solution that no starting material of animal or human origin is used in the fermentation. Elucidation of structure and other characteristics The structure of Everolimus has been fully elucidated using several spectroscopic techniques such as ultraviolet absorption spectroscopy (UV), Infra-red spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance spectroscopy (1 H and 13C NMR), mass spectroscopy, diffractometry (X-ray) and elemental analysis. Related substances An extensive discussion was presented on the related substances. The complex structure of Everolimus allows several possible degradation pathways to occur at various positions of the molecule. Everolimus alone is extremely sensitive to oxidation. By the addition of an antioxidant, the sensitivity to oxidation is significantly reduced (the antioxidant is known to react as a scavenger of peroxide radicals). It is assumed that oxidation of Everolimus proceeds via a radical mechanism. All the requirements set in the current testing instruction valid for Everolimus are justified on the basis of the results obtained during development and manufactured at the production scale.
fda
Everolimus was first approved by Swiss Agency for therapeutic products,Swissmedic on July 18, 2003, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on April 23, 2004, and approved by the U.S. Food and Drug Administration (FDA) on Mar 30, 2009, approved by European Medicine Agency (EMA) on Aug 3, 2009. It was developed and marketed as Certican® by Novartis in SE.
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR). It is indicated for the treatment of renal cell cancer and other tumours and currently used as an immunosuppressant to prevent rejection of organ transplants.
Certican® is available as tablet for oral use, containing 0.25, 0.5 or 0.75 mg of free Everolimus. The recommended dose is 10 mg once daily with or without food for advanced HR+ breast cancer, advanced progressive neuroendocrine tumors, advanced renal cell carcinoma or renal angiomyolipoma with tuberous sclerosis complex. Everolimus, also known as RAD001, is a derivative of the natural macrocyclic lactone sirolimus with immunosuppressant and anti-angiogenic properties. In cells, everolimus binds to the immunophilin FK Binding Protein-12 (FKBP-12) to generate an immunosuppressive complex that binds to and inhibits the activation of the mammalian Target of Rapamycin (mTOR), a key regulatory kinase. Inhibition of mTOR activation results in the inhibition of T lymphocyte activation and proliferation associated with antigen and cytokine (IL-2, IL-4, and IL-15) stimulation and the inhibition of antibody production.
It is marketed by Novartis under the trade names Zortress (USA) and Certican (European Union and other countries) in transplantation medicine, and as Afinitor (general tumours) and Votubia (tumours as a result of TSC) in oncology. Everolimus is also available from Biocon, with the brand name Evertor.
Medical uses
Everolimus is approved for various conditions:
Advanced kidney cancer (US FDA approved in March 2009)[3]
Prevention of organ rejection after renal transplant(US FDA April 2010)[4]
Breast cancer in post-menopausal women with advanced hormone-receptor positive, HER2-negative type cancer, in conjunction with exemestane (US FDA July 2012)[7]
Prevention of organ rejection after liver transplant(Feb 2013)
Progressive, well-differentiated non-functional, neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease (US FDA February 2016).[8]
Tuberous sclerosis complex-associated partial-onset seizures for adult and pediatric patients aged 2 years and older. (US FDA April 2018).[9]
UK National Health Service
NHS England has been criticised for delays in deciding on a policy for the prescription of everolimus in the treatment of Tuberous Sclerosis. 20 doctors addressed a letter to the board in support of the charity Tuberous Scelerosis Association saying ” around 32 patients with critical need, whose doctors believe everolimus treatment is their best or only option, have no hope of access to funding. Most have been waiting many months. Approximately half of these patients are at imminent risk of a catastrophic event (renal bleed or kidney failure) with a high risk of preventable death.”[10] In May 2015 it was reported that Luke Henry and Stephanie Rudwick, the parents of a child suffering from Tuberous Sclerosis were trying to sell their home in Brighton to raise £30,000 to pay for treatment for their daughter Bethany who has tumours on her brain, kidneys and liver and suffers from up to 50 epileptic fits a day.[11]
Interim phase III trial results in 2011 showed that adding Afinitor (everolimus) to exemestane therapy against advanced breast cancer can significantly improve progression-free survival compared with exemestane therapy alone.[14]
A study published in 2012, shows that everolimus sensitivity varies between patients depending on their tumor genomes.[15] A group of patients with advanced metastasic bladder carcinoma (NCT00805129) [16] treated with everolimus revealed a single patient who had a complete response to everolimus treatment for 26 months. The researchers sequenced the genome of this patient and compared it to different reference genomes and to other patients’ genomes. They found that mutations in TSC1 led to a lengthened duration of response to everolimus and to an increase in the time to cancer recurrence. The mutated TSC1 apparently had made these tumors vulnerable to treatment with everolimus.[medical citation needed]
A phase 2a randomized, placebo-controlled everolimus clinical trial published in 2014 showed that everolimus improved the response to an influenza vaccine by 20% in healthy elderly volunteers.[17] A phase 2a randomized, placebo-controlled clinical trial published in 2018 showed that everolimus in combination with dactolisib decreased the rate of reported infections in an elderly population.[17]
Mechanism
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex.[18] This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2 positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types.[medical citation needed] Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.[18]
TSC1 and TSC2, the genes involved in tuberous sclerosis, act as tumor suppressor genes by regulating mTORC1 activity. Thus, either the loss or inactivation of one of these genes lead to the activation of mTORC1.[20]
Everolimus binds to its protein receptor FKBP12, which directly interacts with mTORC1, inhibiting its downstream signaling. As a consequence, mRNAs that code for proteins implicated in the cell cycle and in the glycolysis process are impaired or altered, and tumor growth is inhibited.[20]
Adverse reactions
A trial using 10 mg/day in patients with NETs of GI or lung origin reported “Everolimus was discontinued for adverse reactions in 29% of patients and dose reduction or delay was required in 70% of everolimus-treated patients. Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). The most common adverse reactions (incidence greater than or equal to 30%) were stomatitis, infections, diarrhea, peripheral edema, fatigue and rash. The most common blood abnormalities found (incidence greater than or equal to 50%) were anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase (AST) and fasting hyperglycemia.”.[8]
Role in heart transplantation
Everolimus may have a role in heart transplantation, as it has been shown to reduce chronic allograft vasculopathy in such transplants. It also may have a similar role to sirolimus in kidney and other transplants.[21]
Role in liver transplantation
Although, sirolimus had generated fears over use of m-TOR inhibitors in liver transplantation recipients, due to possible early hepatic artery thrombosis and graft loss, use of everolimus in the setting of liver transplantation is promising. Jeng et al.,[22] in their study of 43 patients, concluded the safety of everolimus in the early phase after living donor liver transplantation. In their study, no hepatic artery thrombosis or wound infection was noted. Also, a possible role of everolimus in reducing the recurrence of hepatocellular carcinoma after liver transplantation was correlated. A target trough level of 3 ng/mL at 3 months was shown to be beneficial in recipients with pre-transplant renal dysfunction. In their study, 6 of 9 renal failure patients showed significant recovery of renal function, whereas 3 showed further deterioration, one of whom required hemodialysis.[23] Recently published report by Thorat et al. showed a positive impact on hepatocellular carcinoma (HCC) when everolimus was used as primary immunosuppression starting as early as first week after living donor liver transplantation (LDLT) surgery.[24] In their retrospective and prospective analysis at China Medical University Hospital in Taiwan, the study cohort (n=66) was divided in two groups depending upon the postoperative immunosuppression. Group A: HCC patients that received Everolimus + Tacrolimus based immunosuppressive regimen (n=37). Group B: HCC patients that received standard Tacrolimus based immunosuppressive regimen without everolimus (n=29). The target trough level for EVR was 3 to 5 ng/ml while for TAC it was 8–10 ng/ml. The 1-year, 3-year and 4-year overall survival achieved for Group A patients (Everolimus group) was 94.95%, 86.48% and 86.48%, respectively while for Group B patients it was 82.75%, 68.96%, and 62.06%, respectively (p=0.0217). The first 12-month report of ongoing Everolimus multicenter prospective trial in LDLT (H2307 trial), Jeng LB et al. have shown a 0% recurrence of HCC in everolimus group at 12 months.[25] Jeng LB concluded that an early introduction of everolimus + reduced tacrolimus was non-inferior to standard tacrolimus in terms of efficacy and renal function at 12 months, with HCC recurrence only in tacrolimus control patients.
Use in vascular stents
Everolimus is used in drug-eluting coronary stents as an immunosuppressant to prevent restenosis. Abbott Vascular produce an everolimus-eluting stent (EES) called Xience Alpine. It utilizes the Multi-Link Vision cobalt chromium stent platform and Novartis’ everolimus. The product is widely available globally including the US, the European Union, and Asia-Pacific (APAC) countries. Boston Scientific also market EESes, recent offerings being Promus Elite and Synergy.[citation needed]
Use in aging
Inhibition of mTOR, the molecular target of everolimus, extends the lifespan of model organisms including mice,[26] and mTOR inhibition has been suggested as an anti-aging therapy. Everolimus was used in a clinical trial by Novartis, and short-term treatment was shown to enhance the response to the influenza vaccine in the elderly, possible by reversing immunosenescence.[27] Everolimus treatment of mice results in reduced metabolic side effects compared to sirolimus.[18]Route 1
Reference:1. US5665772A.
2. Drug. Future1999, 24, 22-29.Route 2
Reference:1. WO2014203185A1.Route 3
Reference:1. WO2012103959A1.Route 4
Reference:1. CN102731527A.
SYN
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 2
Synthetic Reference
Polymer compositions containing a macrocyclic triene compound; Shulze, John E.; Betts, Ronald E.; Savage, Douglas R.; Assignee Sun Bow Co., Ltd., Bermuda; Sun Biomedical Ltd. 2003; Patent Information; Nov 06, 2003; WO 2003090684 A2
SYN 3
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 4
Synthetic Reference
Zabudkin, Oleksandr; Schickaneder, Christian; Matviienko, Iaroslav; Sypchenko, Volodymyr. Method for the synthesis of rapamycin derivatives. Assignee Synbias Pharma AG, Switz. EP 3109250. (2016).
SYN 5
Synthetic Reference
Lu, Shiyong; Zhang, Xiaotian; Chen, Haohan; Ye, Weidong. Preparation of sirolimus 40-ether derivative. Assignee Zhejiang Medicine Co., Ltd. Xinchang Pharmaceutical Factory, Peop. Rep. China. CN 105237549. (2016).
SYN 6
Synthetic Reference
Seo, Jeong U.; Ham, Yun Beom; Kang, Heung Mo; Lee, Gwang Mu; Kim, In Gyu; Kim, Jeong Jin; Park, Ji Su. Preparation of everolimus and synthetic intermediate thereof. Assignee CKD Bio Corp., S. Korea. KR 1529963 (2015).
SYN
EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol.
SYN
J Label Compd Radiopharm 1999,42(1),29
The compound has been obtained biosynthetically by an optimized fermentation process using Streptomyces hygroscopicus mutant RSH 1701 with a complex culture medium were [14C]-labeled (1R,3R,4R)-2,3-dichydroxycyclo-hexanecarboxylic acid (I) and [14C]-labeled (S)-pipecolic acid (II) have been added. This fermentation process yielded [14C]-labeled rapamycin (III), which was finally selectively O-alkylated at the C-40 position with monosilylated ethylene glycol triflate in DMSO/dimethoxyethane.
SYN
The reaction of the labeled acylated (+)-bornane-10,2-sultam (IV) with triethyl phosphite gives the phosphonate (V), which is treated with paraformaldehyde, galvinoxyl and K2CO3 yielding the acrylate derivative (VI). The cyclization of (VI) with butadiene (VII) by means of diethylaluminum chloride and galvinoxyl (as radical scavenger) affords the cyclohexene-carboxamide derivative (VIII), which is hydrolyzed with LiOH in THF/water giving the (1R)-3-cyclohexenecarboxylic acid (IX). The oxidation of (IX) with m-chloroperbenzoic acid and triethylamine in CCl4 yielded regioselectively the hydroxylactone (X), which is finally hydrolyzed with HCl to the labeled intermediate (I).
SYN
The reaction of the labeled acylated (-)-bornane-10,2-sultam (XI) with benzophenone imine (XII) gives the glycylsultam derivative (XIII), which is alkylated with 4-iodobutyl chloride (XIV) by means of butyllithium and DMPU in THF yielding intermediate (XV). The selective hydrolysis of (XV) with HCl affords the omega-chloro-L-norleucine derivative (XVI), which is cyclized by means of tetrabutylammonium fluoride and DIEA in hot acetonitrile giving the (2S)-piperidyl derivative (XVII). Finally, this compound is hydrolyzed with LiOH in THF/water to the labeled intermediate (II).
clipRapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:
See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.
(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):
According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.
(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).
Only 8% yield of the corresponding compound (4B)
and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),
It is a derivative of sirolimus of formula III),
and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress (USA) and Certican (Europe and other countries) in transplantation medicine, and Afinitor in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV),
wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),
was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Sche
Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2- Hydroxy)ethyl rapamycin] of formula (I). However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),
as the alkylation agent. The overall yield reported was 25%. International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),
wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus. European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step. The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity
Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I) Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC. Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS- ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to – 25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol- triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.
Patent
Publication numberPriority datePublication dateAssigneeTitleUS5665772A *1992-10-091997-09-09Sandoz Ltd.O-alkylated rapamycin derivatives and their use, particularly as immunosuppressantsEP1518517A2 *2002-04-242005-03-30Sun Biomedical, Ltd.Drug-delivery endovascular stent and method for treating restenosisWO2012103960A12011-02-042012-08-09Synthon BvProcess for making trisubstituted silyloxyethyl triflatesCN102786534A2012-05-252012-11-21上海现代制药股份有限公司Preparation method of everolimusCN103788114A *2012-10-312014-05-14江苏汉邦科技有限公司Preparation method for everolimusEP3166950A12014-08-042017-05-17Cipla LimitedProcess for the synthesis of everolimus and intermediates thereof
CN107417718A *2017-08-182017-12-01常州兰陵制药有限公司The preparation method of everolimus intermediateUS9938297B22014-08-042018-04-10Cipia LimitedProcess for the synthesis of everolimus and intermediates thereofCN108676014A *2018-06-152018-10-19国药集团川抗制药有限公司The method for purifying the method for everolimus intermediate and preparing everolimus
Ascomycins and rapamycins The ascomycin tacrolimus (44, FK-506) and the two rapamycins sirolimus (45, rapamycin) and everolimus (46) are macrolides that contain 21- and 29-membered macrocyclic rings, respectively (Figure 7).[3] Their MWs range from just over 800 Da for tacrolimus (44) to >900 Da for sirolimus (45) and everolimus (46) and they have >10 HBAs. Like other natural product derived drugs in bRo5 space, they are above average complexity (SMCM 119–134) due to their 14–15 chiral centres. All three are immunosuppressants that are mainly used to prevent rejection of transplanted organs. They bind to overlapping, but slightly different parts of a shallow pocket at the surface of the immunophilin FK506 binding protein (FKBP12, Figure 8 A). Whereas tacrolimus (44) only binds in the pocket on FKBP12 (Figure 8 B),[67] sirolimus (45) and everolimus (46) promote binding of mammalian target of rapamycin (mTOR) so that they bind in a groove formed by FKBP12 and mTOR (Figure 8 C).[68] The complex between tacrolimus (44) and FKBP12 inhibits calcineurin, which results in reduced production of interleukin-2 and inactivation of T cells. Formation of the ternary complexes between FKBP12, sirolimus (45) [or everolimus (46)] and mTOR inhibits mTOR, which arrests growth of T lymphocytes by reducing their sensitivity to interleukin 2. Both tacrolimus (44) and sirolimus (45) have low (15–20 %) and variable bioavailabilities, whereas the bioavailability of everolimus (46) has been increased somewhat as compared to sirolimus (45).[3] Tacrolimus (44) was isolated from Streptomyces tsukubaensis in 1987,[69, 70] while sirolimus (45) was first identified from a Streptomycete strain found in a soil sample from Easter Island.[71] Later it was also isolated from fermentation of another Streptomycete strain.[72, 73] Both drugs are now produced through fermentation.[74, 75] Sirolimus suffers from low bioavailability as well as toxicity, and semi-synthetic derivatives were therefore prepared to minimise these issues. This led to the discovery of everolimus (46), synthesised by selective alkylation of one of the two secondary hydroxyl groups of sirolimus (45) with 2-(tert-butyldimethylsilyl)oxyethyltriflate followed by silyl ether deprotection with HCl (Scheme 8).[76, 77]
Figure 7. Structures of the ascomycin tacrolimus (44) and the rapamycins sirolimus (45) and everolimus (46) that are used mainly to prevent rejection of organ transplants.
[67] G. D. Van Duyne, R. F. Standaert, P. A. Karplus, S. L. Schreiber, J. Clardy, Science 1991, 252, 839 – 842. [68] A. M. Marz, A.-K. Fabian, C. Kozany, A. Bracher, F. Hausch, Mol. Cell. Biol. 2013, 33, 1357 – 1367.
[69] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249 – 1255. [70] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T. Taga, J. Am. Chem. Soc. 1987, 109, 5031 – 5033. [71] C. Vzina, A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721 – 726. [72] S. N. Sehgal, H. Baker, C. Vzina, J. Antibiot. 1975, 28, 727 – 732. [73] S. N. Sehgal, T. M. Blazekovic, C. Vzina, 1975, US3929992A. [74] C. Barreiro, M. Mart nez-Castro, Appl. Microbiol. Biotechnol. 2014, 98, 497 – 507. [75] S. R. Park, Y. J. Yoo, Y.-H. Ban, Y. J. Yoon, J. Antibiot. 2010, 63, 434 – 441. [76] F. Navarro, S. Petit, G. Stone, 2007, US20020032213A1. [77] S. Cottens, R. Sedrani, 1997, US5665772A.
clip
Ferreting out why some cancer drugs struggle to shrink tumors
Study shows how stopping one enzyme could help drugs treat an important class of cancers more effectively
In several types of cancer, including most cases of breast cancer, a cell-signaling network called the PI3K pathway is overactive. Drug designers have tried to quiet this pathway to kill cancer, but they haven’t had much success and, more frustratingly, haven’t understood why the problem is so hard to solve.
“There have been more than 200 clinical trials with experimental drugs that target the PI3K pathway, and probably more than $1 billion invested,” says Sourav Bandyopadhyay of the University of California, San Francisco. Just a handful of drugs have been approved by the U.S. FDA and one, Novartis’s Afinitor (everolimus), deters cancer growth but doesn’t shrink tumors, and it prolongs patient survival only a few months.
Bandyopadhyay, his UCSF colleague John D. Gordan, and coworkers used a proteomics approach to ferret out why previous attempts to target the PI3K pathway have had limited success and, using that information, devised and tested a possible fix (Nat. Chem. Biol. 2018, DOI: 10.1038/s41589-018-0081-9).
The stubborn pathway involves a series of kinases—enzymes that modify other proteins by adding phosphate groups—starting with one called PI3K. Overactivation of the pathway produces the transcription factor MYC, which turns on protein synthesis and can spark cancer growth.
The UCSF team used kinase-affinity beads and tandem mass spectrometry to survey all kinases active in breast cancer cells before and after treatment with a variety of cancer drugs. The team studied this so-called kinome to look for kinases associated with the cells’ tendency to resist drug treatments.
The researchers found that a kinase called AURKA undermines everolimus and other pathway-targeted drugs by reversing their effects. While the drugs try to turn off the PI3K pathway, AURKA, activated separately by other pathways, keeps the PI3K pathway turned on. To add insult to injury, MYC boosts AURKA production, maintaining a plentiful supply of the drug spoiler.
When the researchers coadministered everolimus with the AURKA inhibitor MLN8237, also called alisertib, everolimus could inhibit the PI3K pathway as it was designed to do, without interference. The combination treatment killed most types of cancer cells in culture and shrank tumors in mice with breast cancer, whereas everolimus alone permitted slow tumor growth to continue.
^ Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sørensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P (August 2003). “Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients”. The New England Journal of Medicine. 349 (9): 847–58. doi:10.1056/NEJMoa022171. PMID12944570.
^ Jeng LB, Thorat A, Hsieh YW, Yang HR, Yeh CC, Chen TH, Hsu SC, Hsu CH (April 2014). “Experience of using everolimus in the early stage of living donor liver transplantation”. Transplantation Proceedings. 46 (3): 744–8. doi:10.1016/j.transproceed.2013.11.068. PMID24767339.
EverolimusCAS Registry Number: 159351-69-6CAS Name: 42-O-(2-Hydroxyethyl)rapamycinAdditional Names: 40-O-(2-hydroxyethyl)rapamycinManufacturers’ Codes: RAD-001; SDZ RADTrademarks: Certican (Novartis)Molecular Formula: C53H83NO14Molecular Weight: 958.22Percent Composition: C 66.43%, H 8.73%, N 1.46%, O 23.38%Literature References: Macrolide immunosuppressant; derivative of rapamycin, q.v. Inhibits cytokine-mediated lymphocyte proliferation. Prepn: S. Cottens, R. Sedrani, WO9409010; eidem, US5665772 (1994, 1997 both to Sandoz). Pharmacology: W. Schuler et al., Transplantation64, 36 (1997). Whole blood determn by LC/MS: N. Brignol et al., Rapid Commun. Mass Spectrom.15, 898 (2001); by HPLC: S. Baldelli et al., J. Chromatogr. B816, 99 (2005). Clinical pharmacokinetics in combination with cyclosporine: J. M. Kovarik et al., Clin. Pharmacol. Ther.69, 48 (2001). Clinical study in prevention of cardiac-allograft vasculopathy: H. J. Eisen et al.,N. Engl. J. Med.349, 847 (2003). Review: F. J. Dumont et al., Curr. Opin. Invest. Drugs2, 1220-1234 (2001); B. Nashan, Ther. Drug Monit.24, 53-58 (2002).Therap-Cat: Immunosuppressant.Keywords: Immunosuppressant.эверолимус[Russian][INN]إيفيروليموس[Arabic][INN]依维莫司[Chinese][INN]Trade Name:Certican® / Zortress® / Afinitor®MOA:mTOR inhibitorIndication:Rejection of organ transplantation; Renal cell carcinoma; Advanced renal cell carcinoma (RCC); Advanced breast cancer; Pancreatic cancer; Renal angiomyolipoma; Tuberous sclerosis complex (TSC); Rejection in heart transplantation; Rejection of suppression renal transplantation; Subependymal giant cell astrocytoma; neuroendocrine tumors (NET); Advanced gastrointestinal tumorsStatus:ApprovedCompany:Novartis (Originator)Sales:$1,942 Million (Y2015); $1,902 Million (Y2014); $1,558 Million (Y2013); $1,007 Million (Y2012); $630 Million (Y2011);ATC Code:L04AA18Approved Countries or Area
Rejection of organ transplantation, Renal cell carcinoma
Tablet
0.25 mg/0.5 mg/0.75 mg
Novartis
clip
Active Substance The active substance Everolimus is a hydroxyethyl derivative of rapamycin, which is a macrolide, isolated from the micro-organism Streptomyces hygroscopicus. The guideline, impurities in new active substances ICHQ 3A (R), does not apply to active substance of fermented origin. Everolimus (INN) or 42-O-(2-hydroxyethyl)-rapamycin (chemical name) or C5 3H8 3N O1 4 has been fully described. The molecule is amorphous and is stabilised with an antioxidant. Its physico-chemical properties including parameters such as solubility, pH, specific rotation, potential polymorphism and potential isomerism have been fully characterised. Everolimus is a white to faintly yellow amorphous powder. It is almost insoluble in water, is unstable at temperatures above 25 °C and is sensitive to light. In addition, possible isomerism has been investigated. Everolimus contains 15 asymmetric carbon atoms and 4 substituted double bonds. The configuration of the asymmetric carbon atoms and the double bonds is guaranteed by the microbial origin of Rapamycin. The configuration is not affected by the chemical synthesis. Polymorphism has been comprehensively discussed and it was demonstrated that the molecule domain remains amorphous.
Synthesis of Everolimus The manufacturing process consists of four main steps, (1) fermentation, (2) extraction of rapamycin from the fermentation broth, (3) chemical modification of rapamycin starting material, (4) purification of crude everolimus and stabilisation with BHT. The choice of the stabilizer has been sufficiently explained and justified by experimental results. Interactions products of Everolimus and the antioxidant were not detected, or were below detection limit. Rapamycin, obtained by a fermentation process, was used as the starting material. Reaction conditions and the necessary in-process controls are described in detail. Adequate specifications for starting materials and isolated intermediates and descriptions of the test procedures have been submitted. Control of the quality of solvents, reagents and auxiliary materials used in the synthesis has been adequately documented. It is stated by the manufacturer of rapamycin solution that no starting material of animal or human origin is used in the fermentation. Elucidation of structure and other characteristics The structure of Everolimus has been fully elucidated using several spectroscopic techniques such as ultraviolet absorption spectroscopy (UV), Infra-red spectroscopy (FT-IR), proton and carbon nuclear magnetic resonance spectroscopy (1 H and 13C NMR), mass spectroscopy, diffractometry (X-ray) and elemental analysis. Related substances An extensive discussion was presented on the related substances. The complex structure of Everolimus allows several possible degradation pathways to occur at various positions of the molecule. Everolimus alone is extremely sensitive to oxidation. By the addition of an antioxidant, the sensitivity to oxidation is significantly reduced (the antioxidant is known to react as a scavenger of peroxide radicals). It is assumed that oxidation of Everolimus proceeds via a radical mechanism. All the requirements set in the current testing instruction valid for Everolimus are justified on the basis of the results obtained during development and manufactured at the production scale.
fda
Everolimus was first approved by Swiss Agency for therapeutic products,Swissmedic on July 18, 2003, then approved by Pharmaceuticals and Medicals Devices Agency of Japan (PMDA) on April 23, 2004, and approved by the U.S. Food and Drug Administration (FDA) on Mar 30, 2009, approved by European Medicine Agency (EMA) on Aug 3, 2009. It was developed and marketed as Certican® by Novartis in SE.
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR). It is indicated for the treatment of renal cell cancer and other tumours and currently used as an immunosuppressant to prevent rejection of organ transplants.
Certican® is available as tablet for oral use, containing 0.25, 0.5 or 0.75 mg of free Everolimus. The recommended dose is 10 mg once daily with or without food for advanced HR+ breast cancer, advanced progressive neuroendocrine tumors, advanced renal cell carcinoma or renal angiomyolipoma with tuberous sclerosis complex. Everolimus, also known as RAD001, is a derivative of the natural macrocyclic lactone sirolimus with immunosuppressant and anti-angiogenic properties. In cells, everolimus binds to the immunophilin FK Binding Protein-12 (FKBP-12) to generate an immunosuppressive complex that binds to and inhibits the activation of the mammalian Target of Rapamycin (mTOR), a key regulatory kinase. Inhibition of mTOR activation results in the inhibition of T lymphocyte activation and proliferation associated with antigen and cytokine (IL-2, IL-4, and IL-15) stimulation and the inhibition of antibody production.
It is marketed by Novartis under the trade names Zortress (USA) and Certican (European Union and other countries) in transplantation medicine, and as Afinitor (general tumours) and Votubia (tumours as a result of TSC) in oncology. Everolimus is also available from Biocon, with the brand name Evertor.
Medical uses
Everolimus is approved for various conditions:
Advanced kidney cancer (US FDA approved in March 2009)[3]
Prevention of organ rejection after renal transplant(US FDA April 2010)[4]
Breast cancer in post-menopausal women with advanced hormone-receptor positive, HER2-negative type cancer, in conjunction with exemestane (US FDA July 2012)[7]
Prevention of organ rejection after liver transplant(Feb 2013)
Progressive, well-differentiated non-functional, neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease (US FDA February 2016).[8]
Tuberous sclerosis complex-associated partial-onset seizures for adult and pediatric patients aged 2 years and older. (US FDA April 2018).[9]
UK National Health Service
NHS England has been criticised for delays in deciding on a policy for the prescription of everolimus in the treatment of Tuberous Sclerosis. 20 doctors addressed a letter to the board in support of the charity Tuberous Scelerosis Association saying ” around 32 patients with critical need, whose doctors believe everolimus treatment is their best or only option, have no hope of access to funding. Most have been waiting many months. Approximately half of these patients are at imminent risk of a catastrophic event (renal bleed or kidney failure) with a high risk of preventable death.”[10] In May 2015 it was reported that Luke Henry and Stephanie Rudwick, the parents of a child suffering from Tuberous Sclerosis were trying to sell their home in Brighton to raise £30,000 to pay for treatment for their daughter Bethany who has tumours on her brain, kidneys and liver and suffers from up to 50 epileptic fits a day.[11]
Interim phase III trial results in 2011 showed that adding Afinitor (everolimus) to exemestane therapy against advanced breast cancer can significantly improve progression-free survival compared with exemestane therapy alone.[14]
A study published in 2012, shows that everolimus sensitivity varies between patients depending on their tumor genomes.[15] A group of patients with advanced metastasic bladder carcinoma (NCT00805129) [16] treated with everolimus revealed a single patient who had a complete response to everolimus treatment for 26 months. The researchers sequenced the genome of this patient and compared it to different reference genomes and to other patients’ genomes. They found that mutations in TSC1 led to a lengthened duration of response to everolimus and to an increase in the time to cancer recurrence. The mutated TSC1 apparently had made these tumors vulnerable to treatment with everolimus.[medical citation needed]
A phase 2a randomized, placebo-controlled everolimus clinical trial published in 2014 showed that everolimus improved the response to an influenza vaccine by 20% in healthy elderly volunteers.[17] A phase 2a randomized, placebo-controlled clinical trial published in 2018 showed that everolimus in combination with dactolisib decreased the rate of reported infections in an elderly population.[17]
Mechanism
Compared with the parent compound rapamycin, everolimus is more selective for the mTORC1 protein complex, with little impact on the mTORC2 complex.[18] This can lead to a hyper-activation of the kinase AKT via inhibition on the mTORC1 negative feedback loop, while not inhibiting the mTORC2 positive feedback to AKT. This AKT elevation can lead to longer survival in some cell types.[medical citation needed] Thus, everolimus has important effects on cell growth, cell proliferation and cell survival.
Additionally, mTORC2 is believed to play an important role in glucose metabolism and the immune system, suggesting that selective inhibition of mTORC1 by drugs such as everolimus could achieve many of the benefits of rapamycin without the associated glucose intolerance and immunosuppression.[18]
TSC1 and TSC2, the genes involved in tuberous sclerosis, act as tumor suppressor genes by regulating mTORC1 activity. Thus, either the loss or inactivation of one of these genes lead to the activation of mTORC1.[20]
Everolimus binds to its protein receptor FKBP12, which directly interacts with mTORC1, inhibiting its downstream signaling. As a consequence, mRNAs that code for proteins implicated in the cell cycle and in the glycolysis process are impaired or altered, and tumor growth is inhibited.[20]
Adverse reactions
A trial using 10 mg/day in patients with NETs of GI or lung origin reported “Everolimus was discontinued for adverse reactions in 29% of patients and dose reduction or delay was required in 70% of everolimus-treated patients. Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). The most common adverse reactions (incidence greater than or equal to 30%) were stomatitis, infections, diarrhea, peripheral edema, fatigue and rash. The most common blood abnormalities found (incidence greater than or equal to 50%) were anemia, hypercholesterolemia, lymphopenia, elevated aspartate transaminase (AST) and fasting hyperglycemia.”.[8]
Role in heart transplantation
Everolimus may have a role in heart transplantation, as it has been shown to reduce chronic allograft vasculopathy in such transplants. It also may have a similar role to sirolimus in kidney and other transplants.[21]
Role in liver transplantation
Although, sirolimus had generated fears over use of m-TOR inhibitors in liver transplantation recipients, due to possible early hepatic artery thrombosis and graft loss, use of everolimus in the setting of liver transplantation is promising. Jeng et al.,[22] in their study of 43 patients, concluded the safety of everolimus in the early phase after living donor liver transplantation. In their study, no hepatic artery thrombosis or wound infection was noted. Also, a possible role of everolimus in reducing the recurrence of hepatocellular carcinoma after liver transplantation was correlated. A target trough level of 3 ng/mL at 3 months was shown to be beneficial in recipients with pre-transplant renal dysfunction. In their study, 6 of 9 renal failure patients showed significant recovery of renal function, whereas 3 showed further deterioration, one of whom required hemodialysis.[23] Recently published report by Thorat et al. showed a positive impact on hepatocellular carcinoma (HCC) when everolimus was used as primary immunosuppression starting as early as first week after living donor liver transplantation (LDLT) surgery.[24] In their retrospective and prospective analysis at China Medical University Hospital in Taiwan, the study cohort (n=66) was divided in two groups depending upon the postoperative immunosuppression. Group A: HCC patients that received Everolimus + Tacrolimus based immunosuppressive regimen (n=37). Group B: HCC patients that received standard Tacrolimus based immunosuppressive regimen without everolimus (n=29). The target trough level for EVR was 3 to 5 ng/ml while for TAC it was 8–10 ng/ml. The 1-year, 3-year and 4-year overall survival achieved for Group A patients (Everolimus group) was 94.95%, 86.48% and 86.48%, respectively while for Group B patients it was 82.75%, 68.96%, and 62.06%, respectively (p=0.0217). The first 12-month report of ongoing Everolimus multicenter prospective trial in LDLT (H2307 trial), Jeng LB et al. have shown a 0% recurrence of HCC in everolimus group at 12 months.[25] Jeng LB concluded that an early introduction of everolimus + reduced tacrolimus was non-inferior to standard tacrolimus in terms of efficacy and renal function at 12 months, with HCC recurrence only in tacrolimus control patients.
Use in vascular stents
Everolimus is used in drug-eluting coronary stents as an immunosuppressant to prevent restenosis. Abbott Vascular produce an everolimus-eluting stent (EES) called Xience Alpine. It utilizes the Multi-Link Vision cobalt chromium stent platform and Novartis’ everolimus. The product is widely available globally including the US, the European Union, and Asia-Pacific (APAC) countries. Boston Scientific also market EESes, recent offerings being Promus Elite and Synergy.[citation needed]
Use in aging
Inhibition of mTOR, the molecular target of everolimus, extends the lifespan of model organisms including mice,[26] and mTOR inhibition has been suggested as an anti-aging therapy. Everolimus was used in a clinical trial by Novartis, and short-term treatment was shown to enhance the response to the influenza vaccine in the elderly, possible by reversing immunosenescence.[27] Everolimus treatment of mice results in reduced metabolic side effects compared to sirolimus.[18]Route 1
Reference:1. US5665772A.
2. Drug. Future1999, 24, 22-29.Route 2
Reference:1. WO2014203185A1.Route 3
Reference:1. WO2012103959A1.Route 4
Reference:1. CN102731527A.
SYN
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 2
Synthetic Reference
Polymer compositions containing a macrocyclic triene compound; Shulze, John E.; Betts, Ronald E.; Savage, Douglas R.; Assignee Sun Bow Co., Ltd., Bermuda; Sun Biomedical Ltd. 2003; Patent Information; Nov 06, 2003; WO 2003090684 A2
SYN 3
Synthetic Reference
Wang, Feng. Everolimus intermediate and preparation method thereof. Assignee Shanghai Institute of Pharmaceutical Industry, Peop. Rep. China; China State Institute of Pharmaceutical Industry. CN 109776570. (2019).
SYN 4
Synthetic Reference
Zabudkin, Oleksandr; Schickaneder, Christian; Matviienko, Iaroslav; Sypchenko, Volodymyr. Method for the synthesis of rapamycin derivatives. Assignee Synbias Pharma AG, Switz. EP 3109250. (2016).
SYN 5
Synthetic Reference
Lu, Shiyong; Zhang, Xiaotian; Chen, Haohan; Ye, Weidong. Preparation of sirolimus 40-ether derivative. Assignee Zhejiang Medicine Co., Ltd. Xinchang Pharmaceutical Factory, Peop. Rep. China. CN 105237549. (2016).
SYN 6
Synthetic Reference
Seo, Jeong U.; Ham, Yun Beom; Kang, Heung Mo; Lee, Gwang Mu; Kim, In Gyu; Kim, Jeong Jin; Park, Ji Su. Preparation of everolimus and synthetic intermediate thereof. Assignee CKD Bio Corp., S. Korea. KR 1529963 (2015).
SYN
EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol.
SYN
J Label Compd Radiopharm 1999,42(1),29
The compound has been obtained biosynthetically by an optimized fermentation process using Streptomyces hygroscopicus mutant RSH 1701 with a complex culture medium were [14C]-labeled (1R,3R,4R)-2,3-dichydroxycyclo-hexanecarboxylic acid (I) and [14C]-labeled (S)-pipecolic acid (II) have been added. This fermentation process yielded [14C]-labeled rapamycin (III), which was finally selectively O-alkylated at the C-40 position with monosilylated ethylene glycol triflate in DMSO/dimethoxyethane.
SYN
The reaction of the labeled acylated (+)-bornane-10,2-sultam (IV) with triethyl phosphite gives the phosphonate (V), which is treated with paraformaldehyde, galvinoxyl and K2CO3 yielding the acrylate derivative (VI). The cyclization of (VI) with butadiene (VII) by means of diethylaluminum chloride and galvinoxyl (as radical scavenger) affords the cyclohexene-carboxamide derivative (VIII), which is hydrolyzed with LiOH in THF/water giving the (1R)-3-cyclohexenecarboxylic acid (IX). The oxidation of (IX) with m-chloroperbenzoic acid and triethylamine in CCl4 yielded regioselectively the hydroxylactone (X), which is finally hydrolyzed with HCl to the labeled intermediate (I).
SYN
The reaction of the labeled acylated (-)-bornane-10,2-sultam (XI) with benzophenone imine (XII) gives the glycylsultam derivative (XIII), which is alkylated with 4-iodobutyl chloride (XIV) by means of butyllithium and DMPU in THF yielding intermediate (XV). The selective hydrolysis of (XV) with HCl affords the omega-chloro-L-norleucine derivative (XVI), which is cyclized by means of tetrabutylammonium fluoride and DIEA in hot acetonitrile giving the (2S)-piperidyl derivative (XVII). Finally, this compound is hydrolyzed with LiOH in THF/water to the labeled intermediate (II).
clipRapamycin is a known macrolide antibiotic produced by Streptomvces hvgroscopicus. having the structure depicted in Formula A:
See, e.g., McAlpine, J.B., et al., J. Antibiotics (1991) 44: 688; Schreiber, S.L., et al., J. Am. Chem. Soc. (1991) J_13: 7433‘- US Patent No. 3 929 992. Rapamycin is an extremely potent immunosuppressant and has also been shown to have antitumor and antifungal activity. Its utility as a pharmaceutical, however, is restricted by its very low and variable bioavailabiiity as well as its high toxicity. Moreover, rapamycin is highly insoluble, making it difficult to formulate stable galenic compositions.
Everolimus, 40-O-(2-hydroxyethyl)-rapamycin of formula (1) is a synthetic derivative of rapamycin (sirolimus) of formula (2), which is produced by a certain bacteria strain and is also pharmaceutically active.
(1) (2)
Everolimus is marketed under the brand name Certican for the prevention of rejection episodes following heart and kidney transplantation, and under the brand name Afinitor for treatment of advanced kidney cancer.
Due to its complicated macrolide chemical structure, everolimus is, similarly as the parent rapamycin, an extremely unstable compound. It is sensitive, in particular, towards oxidation, including aerial oxidation. It is also unstable at temperatures higher than 25°C and at alkaline pH.
Everolimus and a process of making it have been disclosed in WO 94/09010
Synthesis
Alkylation of rapamycin (I) with 2-(tert-butyldimethylsilyloxy)ethyl triflate (II) by means of 2,6-lutidine in hot toluene gives the silylated target compound (III), which is deprotected by means of 1N HCl in methanol (1). (Scheme 21042401a) Manufacturer Novartis AG (CH). References 1. Cottens, S., Sedrani, R. (Sandoz-Refindungen VmbH; Sandoz-Patent GmbH; Sandoz Ltd.). O-Alkylated rapamycin derivatives and their use, particularly as immunosuppressants. EP 663916, EP 867438, JP 96502266, US 5665772, WO 9409010.EP 0663916; EP 0867438; JP 1996502266; JP 1999240884; US 5665772; WO 9409010
(US 5,665,772, EP 663916). The process principle is shown in the scheme below, wherein the abbreviation RAP-OH has been used as an abbreviation for the rapamycin structure of formula (2) above, L is a leaving group and P is a trisubstituted silyl group serving as a OH- protective group.
Specifically, the L- group is a trifluoromethanesulfonate (triflate) group and the protective group P- is typically a tert-butyldimethylsilyloxy- group. Accordingly, the known useful reagent within the above general formula (3) for making everolimus from rapamycin is 2-(tert-butyldimethylsilyloxy)ethyl triflate of formula (3 A):
According to a known synthetic procedure disclosed in Example 8 of WO 94/09010 and in Example 1 of US application 2003/0125800, rapamycin (2) reacts in hot toluene and in the presence of 2,6-lutidine with a molar excess of the compound (3 A), which is charged in several portions, to form the t-butyldimethylsilyl-protected everolimus (4A). This compound is isolated and deprotected by means of IN aqueous HC1 in methanol. Crude everolimus is then purified by column chromatography. Yields were not reported.
(2) (3A) (4A) (1)
In an article of Moenius et al. (J. Labelled Cpd. Radiopharm. 43, 113-120 (2000)), which used the above process for making C14-labelled and tritiated everolimus, a diphenyl- tert.butylsilyloxy -protective group was used as the alkylation agent of formula (3B).
Only 8% yield of the corresponding compound (4B)
and 21% yield of the compound (1) have been reported.
Little is known about the compounds of the general formula (3) and methods of their preparation. The synthesis of the compound (3 A) was disclosed in Example 1 of US application 2003/0125800. It should be noted that specification of the reaction solvent in the key step B of this synthesis was omitted in the disclosure; however, the data about isolation of the product allow for estimation that such solvent is dichloromethane. Similarly also a second article of Moenius et al. (J. Labelled Cpd. Radiopharm.42, 29-41 (1999)) teaches that dichloromethane is the solvent in the reaction.
It appears that the compounds of formula (3) are very reactive, and thus also very unstable compounds. This is reflected by the fact that the yields of the reaction with rapamycine are very low and the compound (3) is charged in high molar extent. Methods how to monitor the reactivity and/or improve the stability of compounds of general formula (3), however, do not exist.
Thus, it would be useful to improve both processes of making compounds of formula (3) and, as well, processes of their application in chemical synthesis.
In a 100 mL flask, Rapamycin (6 g, 6.56 mmol) was dissolved in dimethoxyethane (4.2 ml) and toluene (24 ml) to give a white suspension and the temperature was raised to 70°C. After 20 min, N,N-diisopropylethylamine (4.56 ml, 27.6 mmol) and 2-((2,3-dimethylbutan-2- yl)dimethylsilyloxy)ethyl trifluoromethanesulfonate (8.83 g, 26.3 mmol) were added in 2 portions with a 2 hr interval at 70°C. The mixture was stirred overnight at room temperature, then diluted with EtOAc (40 ml) and washed with sat. NaHC03 (30 ml) and brine (30 ml). The organic layer was dried with Na2S04, filtered and concentrated. The cmde product was chromatographed on a silica gel column (EtOAc/heptane 1/1 ; yield 4.47 g).
Example 7: 40-O-(2-hydroxyethyl)-rapamycin [everolimus]
In a 100 mL flask, 40-O-[2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl]rapamycin (4.47 g, 4.06 mmol) was dissolved in methanol (20 ml) to give a colorless solution. At 0°C, IN aqueous hydrochloric acid (2.0 ml, 2.0 mmol) was added and the mixture was stirred for 90 min. The reaction was followed by TLC (ethyl acetate/n-heptane 3 :2) and HPLC. Then 20 ml of saturated aqueous NaHC03 were added, followed by 20 ml of brine and 80 ml of ethyl acetate. The phases were separated and the organic layer was washed with saturated aqueous NaCl until pH 6/7. The organic layer was dried by Na2S04, filtered and concentrated to yield 3.3 g of the product.
a) 40-O-[2-(t-Butyldimethylsilyl)oxy]ethyl-rapamycin
A solution of 9.14 g (10 mmol) of rapamycin and 4.70 mL (40 mmol) of 2,6-lutidine in 30 mL of toluene is warmed to 60°C and a solution of 6.17 g (20 mmol) of 2-(t-butyldimethylsilyl)oxyethyl triflate and 2.35 mL (20 mmol) of 2,6-lutidine in 20 mL of toluene is added. This mixture is stirred for 1.5h. Then two batches of a solution of 3.08 g (10 mmol) of triflate and 1.2 mL (10 mmol) of 2,6-lutidine in 10 mL of toluene are added in a 1.5h interval. After addition of the last batch, stirring is continued at 60°C for 2h and the resulting brown suspension is filtered. The filtrate is diluted with ethyl acetate and washed with aq. sodium bicarbonate and brine. The organic solution is dried over anhydrous sodium sulfate, filtered and concentrated. The residue is purified by column chromatography on silica gel (40:60 hexane-ethyl acetate) to afford 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin as a white solid: 1H NMR (CDCl3) δ 0.06 (6H, s), 0.72 (1H, dd), 0.90 (9H, s), 1.65 (3H, s), 1.75 (3H, s), 3.02 (1H, m), 3.63 (3H, m), 3.72 (3H, m); MS (FAB) m/z 1094 ([M+Na]+), 1022 ([M-(OCH3+H2O)]+).
b) 40-O-(2-Hydroxy)ethyl-rapamycin
To a stirred, cooled (0°C) solution of 4.5 g (4.2 mmol) of 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl-rapamycin in 20 mL of methanol is added 2 mL of IN HCl. This solution is stirred for 2h and neutralized with aq. sodium bicarbonate. The mixture is extracted with three portions of ethyl acetate. The organic solution is washed with aq.
sodium bicarbonate and brine, dried over anhydrous sodium sulfate, filtered and
concentrated. Purification by column chromatography on silica gel (ethyl acetate) gave the title compound as a white solid:1H NMR (CDCl3) δ 0.72 (1H, dd), 1.65 (3H, s), 1.75 (3H, s), 3.13 (5H, s and m), 3.52-3.91 (8H, m); MS (FAB) m/z 980 ([M+Na]+), 926 ([M-OCH3]+), 908 ([M-(OCH3+H2O)]+), 890 ([M-(OCH3+2H2O)]+), 876 ([M-(2CH3OH+OH)]+), 858 ([M-(OCH3+CH3OH+2H2O)]+).
MBA (rel. IC50) 2.2
IL-6 dep. prol. (rel. IC50) 2.8
MLR (rel. IC50) 3.4
…………………..
synthesis
Everolimus (Everolimus) was synthesized by the Sirolimus (sirolimus, also known as rapamycin Rapamycin) ether from. Sirolimus is from the soil bacterium Streptomyces hygroscopicus isolated metabolites. Activation end sirolimus (triflate, Tf) the other end of the protection (t-butyldimethylsilyl, TBS) of ethylene glycol 1 reaction of 2 , because the hydroxyl group 42 hydroxyl site over the 31-bit resistance is small, so the reaction only occurs in 42. Compound 2under acidic conditions TBS protection is removed everolimus.
Everolimus (RAD-001) is the 40-O- 2-hydroxyethyl)-rapamycin of formula (I),
It is a derivative of sirolimus of formula III),
and works similarly to sirolimus as an inhibitor of mammalian target of rapamycin (mTOR). Everolimus is currently used as an immunosuppressant to prevent rejection of organ transplants and treatment of renal cell cancer and other tumours. It is marketed by Novartis under the tradenames Zortress (USA) and Certican (Europe and other countries) in transplantation medicine, and Afinitor in oncology.
Trisubstituted silyloxyethyltrifluoromethane sulfonates (triflates) of the general formula (IV),
wherein R2, R3 are independently a straight or branched alkyl group, for example C^-Cw alkyl, and/or an aryl group, for example a phenyl group, are important intermediates useful in the synthesis of everolimus.
Everolimus and its process for manufacture using the intermediate 2-(t-butyldimethyl silyl) oxyethyl triflate of formula (IVA),
was first described in US Patent Number 5,665,772. The overall reaction is depicted in Scheme I.
Sche
Everolimus (I)
For the synthesis, firstly sirolimus of formula (III) and 2-(t-butyldimethylsilyl)oxyethyl triflate of formula (IVA) are reacted in the presence of 2,6-Lutidine in toluene at around 60°C to obtain the corresponding 40-O-[2-(t-butyldimethylsilyl)oxy]ethyl rapamycin of formula (I la), which is then deprotected in aqueous hydrochloric acid and converted into crude everolimus [40-O-(2- Hydroxy)ethyl rapamycin] of formula (I). However, this process results in the formation of impure everolimus, which requires purification by column chromatography. The process results in very poor overall yield and purity and thereby the process is not suitable for the commercial scale production of everolimus.
Moenius et al. (I. Labelled Cpd. Radiopharm. 43, 1 13-120 (2000) have disclosed a process to prepare C-14 labelled everolimus using the diphenyltert-butylsilyloxy-protective group of formula (IV B),
as the alkylation agent. The overall yield reported was 25%. International patent application, publication number WO 2012/103960 discloses the preparation of everolimus using the alkylating agent 2-((2,3-dimethylbut-2-yl)dimethylsilyloxy)ethyl triflate of formula (IVC),
wherein the overall yield reported is 52.54%. The process involves a derivatization method based on the reaction of the triflate (IV) with a derivatization agent, which preferably is a secondary aromatic amine, typically N-methylaniline.
International patent application, publication number WO 2012/103959 also discloses the preparation of everolimus using the alkylating agent of formula (IVC). The process is based on a reaction of rapamycin with the compound of formula (IVC) in the presence of a base (such as an aliphatic tertiary amine) to form 40-O-2-(t-hexyldimethylsiloxy)ethylrapamycin, which is subsequently deprotected under acidic conditions to obtain everolimus. European Patent Number 1518517B discloses a process for the preparation of everolimus which employs the triflate compound of formula (IVA), 2-(t-butyldimethyl silyl) oxyethyl triflate. The disclosed process for preparing the compound of formula (IVA) involves a flash chromatography purification step. The compounds of formula (IV) are key intermediates in the synthesis of everolimus. However, they are highly reactive and also very unstable, and their use often results in decomposition during reaction with sirolimus. This is reflected by the fact that the yields of the reaction with sirolimus are very low and the compounds of formula (IV) are charged in high molar extent. Thus it is desirable to develop a process to stabilize compounds of formula (IV) without loss of reactivity
Example 1 :
Step 1 : Preparation of protected everolimus (TBS-everoismus) of formula (Ma) using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in dichloromethane (DCM) (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). To this sirolimus solution, silver acetate (0.018g, 0.000109mol) was added and cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. The reaction was monitored by TLC. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in the next step. HPLC product purity: 60%-85%.
Step 2: Preparation of everolimus of formula (I) Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus (0.8 g). The crude everolimus was further purified by preparative HPLC to yield everolimus of purity >99%.
Example 2:
Step 1 : Preparation of TBS-everoiimus of formula (Ma) without using metal salt, wherein “Pg” is t-butyldimethylsilyl t-butyldimethylsilyloxy ethanol, of formula (VA) (2.8g, 0.016mol) was dissolved in DCM (3 vol) and to this 2,6-Lutidine (3.50 g, 0.0327 mol) was added and the mixture was cooled to -40°C. Thereafter, trifluoromethane sulfonic anhydride (3.59ml, 0.021 mol) was added drop-wise. The mixture was maintained at -40°C for 30 minutes. Sirolimus (0.5g, 0.00054mol) was taken in another flask and dissolved in DCM (1 ml). The solution was cooled to -40°C. The earlier cooled triflate solution was transferred in 3 lots to the sirolimus solution maintaining temperature at -40°C. The reaction mixture was stirred at -40°C further for 15min before which it was slowly warmed to 0°C and further to RT. The reaction mixture was then warmed to 40°C and maintained at this temperature for 3 hours. On completion of reaction, the reaction mixture was diluted with DCM and washed with water and brine. The organic layer was dried over anhydrous sodium sulphate and solvent was removed by vacuum distillation to obtain the title compound, which was directly used in next step. HPLC purity: 10%-20%.
Step 2: Preparation of everolimus of formula (I)
Protected everolimus of formula (I la) obtained in step 1 was dissolved in methanol (10 volumes) and chilled to 0-5° C. To this solution was added drop wise, a solution of 1 N HCI. The pH of the reaction was maintained between 1-3. The temperature of the reaction mixture was raised to 25° C and stirred for 1 hour. After completion of reaction, the reaction mixture was diluted with water (15 volumes) and extracted in ethyl acetate (2X20 volumes). The organic layers were combined and washed with brine, dried over sodium sulphate. The organic layer was distilled off under reduced pressure at 30-35° C, to obtain a crude everolimus which was further purified by preparative HPLC. Example 3:
Preparation of crude Everolimus
Step 1 : Preparation of TBS-ethylene glycol of formula (Va)
Ethylene glycol (1.5L, 26.58 mol) and TBDMS-CI (485g, 3.21 mol) were mixed together with stirring and cooled to 0°C. Triethyl amine (679 ml, 4.83 mol) was then added at 0°C in 30-45 minutes. After addition, the reaction was stirred for 12 hours at 25-30°C for the desired conversion. After completion of reaction, the layers were separated and the organic layer (containing TBS- ethylene glycol) was washed with water (1 L.x2) and brine solution (1 L). The organic layer was then subjected to high vacuum distillation to afford 350g of pure product.
Step 2: Preparation of TBS-glycol-Triflate of formula (IVa)
The reaction was carried out under a nitrogen atmosphere. TBS- ethylene glycol prepared as per step 1 (85.10g, 0.48 mol) and 2, 6-Lutidine (84.28ml, 0.72 mol) were stirred in n-heptane (425ml) to give a clear solution which was then cooled to -15 to – 25°C. Trif!uoromethanesulfonic anhydride (Tf20) (99.74 ml, 0.590 mol) was added drop-wise over a period of 45 minutes to the n-heptane solution (white precipitate starts to form immediately) while maintaining the reaction at -15 to – 25°C. The reaction mixture was kept at temperature between -15 to -25°C for 2 hours. The precipitate generated was filtered off. The filtrate was then evaporated up to ~2 volumes with respect to TBS-ethyiene glycol (~200 ml).
Step 3: Preparation of TBS-evero!imus of formula (Ha)
30g of sirolimus (0,0328 mo!) and toluene (150m!) were stirred together and the temperature was slowly raised to 60-65°C. At this temperature, a first portion of TBS-g!yco!-triflate prepared as per step 2 (100ml) and 2,6-Lutidine (1 1.45ml, 0.086 moles) were added and stirred for 40 min. Further, a second portion of TBS- glycol-triflate (50mi) and 2, 6-Lutidine (19.45ml, 0.138 mol) were added and the reaction was stirred for another 40 min. This was followed by a third portion of TBS- glycol- triflate (50m!) and 2, 6-Lutidine (19.45ml, 0.138 mol), after which the reaction was stirred for further 90 minutes. The reaction was monitored through HPLC to check the conversion of Sirolimus to TBS-everolimus after each addition of TBS-glycol-trifiate. After completion of the reaction, the reaction mixture was diluted with n-heptane (150mi), cooled to room temperature and stirred for another 60 minutes. The precipitated solids were filtered off and the filtrate was washed with deionized water (450 ml x4) followed by brine solution (450ml). The filtrate was subsequently distilled off to afford TBS-everolimus (60-65g) with 60-70% conversion from sirolimus.
Step 4: Preparation of everolimus of formula (I)
TBS-everolimus (65g) obtained in step 3 was dissolved in 300 mi methanol and cooled to 0°C. 1 N HCI was then added to the methanol solution (pH adjusted to 2-3) and stirred for 2 h. After completion of reaction, toluene (360m!) and deionized wafer (360mi) were added to the reaction mixture and the aqueous layer was separated. The organic layer was washed with brine solution (360ml). The organic layer was concentrated to obtain crude everolimus (39g) with an assay content of 30-35%, HPLC purity of 60-65%.
The crude everolimus purified by chromatography to achieve purity more than 99 %.
Patent
Publication numberPriority datePublication dateAssigneeTitleUS5665772A *1992-10-091997-09-09Sandoz Ltd.O-alkylated rapamycin derivatives and their use, particularly as immunosuppressantsEP1518517A2 *2002-04-242005-03-30Sun Biomedical, Ltd.Drug-delivery endovascular stent and method for treating restenosisWO2012103960A12011-02-042012-08-09Synthon BvProcess for making trisubstituted silyloxyethyl triflatesCN102786534A2012-05-252012-11-21上海现代制药股份有限公司Preparation method of everolimusCN103788114A *2012-10-312014-05-14江苏汉邦科技有限公司Preparation method for everolimusEP3166950A12014-08-042017-05-17Cipla LimitedProcess for the synthesis of everolimus and intermediates thereof
CN107417718A *2017-08-182017-12-01常州兰陵制药有限公司The preparation method of everolimus intermediateUS9938297B22014-08-042018-04-10Cipia LimitedProcess for the synthesis of everolimus and intermediates thereofCN108676014A *2018-06-152018-10-19国药集团川抗制药有限公司The method for purifying the method for everolimus intermediate and preparing everolimus
Ascomycins and rapamycins The ascomycin tacrolimus (44, FK-506) and the two rapamycins sirolimus (45, rapamycin) and everolimus (46) are macrolides that contain 21- and 29-membered macrocyclic rings, respectively (Figure 7).[3] Their MWs range from just over 800 Da for tacrolimus (44) to >900 Da for sirolimus (45) and everolimus (46) and they have >10 HBAs. Like other natural product derived drugs in bRo5 space, they are above average complexity (SMCM 119–134) due to their 14–15 chiral centres. All three are immunosuppressants that are mainly used to prevent rejection of transplanted organs. They bind to overlapping, but slightly different parts of a shallow pocket at the surface of the immunophilin FK506 binding protein (FKBP12, Figure 8 A). Whereas tacrolimus (44) only binds in the pocket on FKBP12 (Figure 8 B),[67] sirolimus (45) and everolimus (46) promote binding of mammalian target of rapamycin (mTOR) so that they bind in a groove formed by FKBP12 and mTOR (Figure 8 C).[68] The complex between tacrolimus (44) and FKBP12 inhibits calcineurin, which results in reduced production of interleukin-2 and inactivation of T cells. Formation of the ternary complexes between FKBP12, sirolimus (45) [or everolimus (46)] and mTOR inhibits mTOR, which arrests growth of T lymphocytes by reducing their sensitivity to interleukin 2. Both tacrolimus (44) and sirolimus (45) have low (15–20 %) and variable bioavailabilities, whereas the bioavailability of everolimus (46) has been increased somewhat as compared to sirolimus (45).[3] Tacrolimus (44) was isolated from Streptomyces tsukubaensis in 1987,[69, 70] while sirolimus (45) was first identified from a Streptomycete strain found in a soil sample from Easter Island.[71] Later it was also isolated from fermentation of another Streptomycete strain.[72, 73] Both drugs are now produced through fermentation.[74, 75] Sirolimus suffers from low bioavailability as well as toxicity, and semi-synthetic derivatives were therefore prepared to minimise these issues. This led to the discovery of everolimus (46), synthesised by selective alkylation of one of the two secondary hydroxyl groups of sirolimus (45) with 2-(tert-butyldimethylsilyl)oxyethyltriflate followed by silyl ether deprotection with HCl (Scheme 8).[76, 77]
Figure 7. Structures of the ascomycin tacrolimus (44) and the rapamycins sirolimus (45) and everolimus (46) that are used mainly to prevent rejection of organ transplants.
[67] G. D. Van Duyne, R. F. Standaert, P. A. Karplus, S. L. Schreiber, J. Clardy, Science 1991, 252, 839 – 842. [68] A. M. Marz, A.-K. Fabian, C. Kozany, A. Bracher, F. Hausch, Mol. Cell. Biol. 2013, 33, 1357 – 1367.
[69] T. Kino, H. Hatanaka, M. Hashimoto, M. Nishiyama, T. Goto, M. Okuhara, M. Kohsaka, H. Aoki, H. Imanaka, J. Antibiot. 1987, 40, 1249 – 1255. [70] H. Tanaka, A. Kuroda, H. Marusawa, H. Hatanaka, T. Kino, T. Goto, M. Hashimoto, T. Taga, J. Am. Chem. Soc. 1987, 109, 5031 – 5033. [71] C. Vzina, A. Kudelski, S. N. Sehgal, J. Antibiot. 1975, 28, 721 – 726. [72] S. N. Sehgal, H. Baker, C. Vzina, J. Antibiot. 1975, 28, 727 – 732. [73] S. N. Sehgal, T. M. Blazekovic, C. Vzina, 1975, US3929992A. [74] C. Barreiro, M. Mart nez-Castro, Appl. Microbiol. Biotechnol. 2014, 98, 497 – 507. [75] S. R. Park, Y. J. Yoo, Y.-H. Ban, Y. J. Yoon, J. Antibiot. 2010, 63, 434 – 441. [76] F. Navarro, S. Petit, G. Stone, 2007, US20020032213A1. [77] S. Cottens, R. Sedrani, 1997, US5665772A.
clip
Ferreting out why some cancer drugs struggle to shrink tumors
Study shows how stopping one enzyme could help drugs treat an important class of cancers more effectively
In several types of cancer, including most cases of breast cancer, a cell-signaling network called the PI3K pathway is overactive. Drug designers have tried to quiet this pathway to kill cancer, but they haven’t had much success and, more frustratingly, haven’t understood why the problem is so hard to solve.
“There have been more than 200 clinical trials with experimental drugs that target the PI3K pathway, and probably more than $1 billion invested,” says Sourav Bandyopadhyay of the University of California, San Francisco. Just a handful of drugs have been approved by the U.S. FDA and one, Novartis’s Afinitor (everolimus), deters cancer growth but doesn’t shrink tumors, and it prolongs patient survival only a few months.
Bandyopadhyay, his UCSF colleague John D. Gordan, and coworkers used a proteomics approach to ferret out why previous attempts to target the PI3K pathway have had limited success and, using that information, devised and tested a possible fix (Nat. Chem. Biol. 2018, DOI: 10.1038/s41589-018-0081-9).
The stubborn pathway involves a series of kinases—enzymes that modify other proteins by adding phosphate groups—starting with one called PI3K. Overactivation of the pathway produces the transcription factor MYC, which turns on protein synthesis and can spark cancer growth.
The UCSF team used kinase-affinity beads and tandem mass spectrometry to survey all kinases active in breast cancer cells before and after treatment with a variety of cancer drugs. The team studied this so-called kinome to look for kinases associated with the cells’ tendency to resist drug treatments.
The researchers found that a kinase called AURKA undermines everolimus and other pathway-targeted drugs by reversing their effects. While the drugs try to turn off the PI3K pathway, AURKA, activated separately by other pathways, keeps the PI3K pathway turned on. To add insult to injury, MYC boosts AURKA production, maintaining a plentiful supply of the drug spoiler.
When the researchers coadministered everolimus with the AURKA inhibitor MLN8237, also called alisertib, everolimus could inhibit the PI3K pathway as it was designed to do, without interference. The combination treatment killed most types of cancer cells in culture and shrank tumors in mice with breast cancer, whereas everolimus alone permitted slow tumor growth to continue.
^ Eisen HJ, Tuzcu EM, Dorent R, Kobashigawa J, Mancini D, Valantine-von Kaeppler HA, Starling RC, Sørensen K, Hummel M, Lind JM, Abeywickrama KH, Bernhardt P (August 2003). “Everolimus for the prevention of allograft rejection and vasculopathy in cardiac-transplant recipients”. The New England Journal of Medicine. 349 (9): 847–58. doi:10.1056/NEJMoa022171. PMID12944570.
^ Jeng LB, Thorat A, Hsieh YW, Yang HR, Yeh CC, Chen TH, Hsu SC, Hsu CH (April 2014). “Experience of using everolimus in the early stage of living donor liver transplantation”. Transplantation Proceedings. 46 (3): 744–8. doi:10.1016/j.transproceed.2013.11.068. PMID24767339.
BuspironeCAS Registry Number: 36505-84-7CAS Name: 8-[4-[4-(2-Pyrimidinyl)-1-piperazinyl]butyl]-8-azaspiro[4.5]decane-7,9-dioneMolecular Formula: C21H31N5O2Molecular Weight: 385.50Percent Composition: C 65.43%, H 8.11%, N 18.17%, O 8.30%Literature References: Non-benzodiazepine anxiolytic; 5-hydroxytryptamine (5-HT1) receptor agonist. Prepn: Y. H. Wu et al.,J. Med. Chem.15, 477 (1972); Y. H. Wu, J. W. Rayburn, DE2057845 (1971 to Bristol-Myers); eidem,US3717634 (1973 to Mead-Johnson). Pharmacology: L. E. Allen et al.,Arzneim.-Forsch.24, 917 (1974). Comparison with diazepam in treatment of anxiety: H. L. Goldberg, R. J. Finnerty, Am. J. Psychiatry136, 1184 (1979); A. F. Jacobson et al.,Pharmacotherapy5, 290 (1985). Nonsynergistic effect with alcohol: T. Seppala et al.,Clin. Pharmacol. Ther.32, 201 (1982). Disposition and metabolism: S. Caccia et al.,Xenobiotica13, 147 (1983). Series of articles on chemistry, pharmacology, addictive potential, and clinical trials: J. Clin. Psychiatry43, pp 1-116 (1982); on pharmacology, safety and clinical comparison with clorazepate: Am. J. Med.80, Suppl. 3B, 1-51 (1986). Review of pharmacology and therapeutic efficacy: K. L. Goa, A. Ward, Drugs32, 114-129 (1986). Review: M. W. Jann, Pharmacotherapy8, 100-116 (1988); D. P. Taylor, FASEB J.2, 2445-2452 (1988). Derivative Type: HydrochlorideCAS Registry Number: 33386-08-2Trademarks: Ansial (Vita); Ansiced (Abello); Axoren (Glaxo Wellcome); Bespar (BMS); Buspar (BMS); Buspimen (Menarini); Buspinol (Zdravlje); Buspisal (Lesvi); Narol (Almirall)Molecular Formula: C21H31N5O2.HClMolecular Weight: 421.96Percent Composition: C 59.77%, H 7.64%, N 16.60%, O 7.58%, Cl 8.40%Properties: Crystals from abs ethanol, mp 201.5-202.5°. LD50 i.p. in rats: 136 mg/kg (Allen).Melting point: mp 201.5-202.5°Toxicity data: LD50 i.p. in rats: 136 mg/kg (Allen) Therap-Cat: Anxiolytic.Keywords: Anxiolytic; Arylpiperazines; Serotonin Receptor Agonist.
Buspirone, sold under the brand name Buspar, among others, is a medication primarily used to treat anxiety disorders, particularly generalized anxiety disorder.[9][10] Benefits support its short term use.[11] It has not been found to be effective in treating psychosis.[9] It is taken by mouth, and it may take up to four weeks to have an effect.[9][10]
Buspirone was first made in 1968 and approved for medical use in the United States in 1986.[9][10] It is available as a generic medication.[11] In 2018, it was the 92nd most-commonly prescribed medication in the United States, with more than 8 million prescriptions.[13][14]
Buspirone has no immediate anxiolytic effects, and hence has a delayed onset of action; its full clinical effectiveness may require 2–4 weeks to manifest itself.[20] The drug has been shown to be similarly effective in the treatment of generalized anxiety disorder (GAD) to benzodiazepines including diazepam, alprazolam, lorazepam, and clorazepate.[2] Buspirone is not known to be effective in the treatment of other anxiety disorders besides GAD,[21] although there is some limited evidence that it may be useful in the treatment of social phobia as an adjunct to selective serotonin reuptake inhibitors (SSRIs).[2][22]
SSRI and SNRI antidepressants such as paroxetine and venlafaxine may cause jaw pain/jaw spasm reversible syndrome (although it is not common), and buspirone appears to be successful in treating bruxism on SSRI/SNRI-induced jaw clenching.[25][26]
It is unclear if there is a risk of tardive dyskinesia or other movement disorders with buspirone.[9]
Overdose
Buspirone appears to be relatively benign in cases of single-drug overdose, although no definitive data on this subject appear to be available.[29] In one clinical trial, buspirone was administered to healthy male volunteers at a dosage of 375 mg/day, and produced side effects including nausea, vomiting, dizziness, drowsiness, miosis, and gastric distress.[15][16][18] In early clinical trials, buspirone was given at dosages even as high as 2,400 mg/day, with akathisia, tremor, and muscle rigidity observed.[30] Deliberate overdoses with 250 mg and up to 300 mg buspirone have resulted in drowsiness in about 50% of individuals.[30] One death has been reported in association with 450 mg buspirone together with alprazolam, diltiazem, alcohol, cocaine.[30]
Interactions
Buspirone has been shown in vitro to be metabolized by the enzymeCYP3A4.[8] This finding is consistent with the in vivo interactions observed between buspirone and these inhibitors or inducers of cytochrome P450 3A4 (CYP3A4), among others:[27]
In addition to binding to serotonin receptors, buspirone is an antagonist of the dopamineD2 receptor with weak affinity.[2][35] It preferentially blocks inhibitory presynaptic D2 autoreceptors, and antagonizes postsynaptic D2 receptors only at higher doses.[2] In accordance, buspirone has been found to increase dopaminergicneurotransmission in the nigrostriatal pathway at low doses, whereas at higher doses, postsynaptic D2 receptors are blocked and antidopaminergic effects such as hypoactivity and reduced stereotypy, though notably not catalepsy, are observed in animals.[2] Buspirone has also been found to bind with much higher affinity to the dopamine D3 and D4 receptors, where it is similarly an antagonist.[45]
A major metabolite of buspirone, 1-(2-pyrimidinyl)piperazine (1-PP), occurs at higher circulating levels than buspirone itself and is known to act as a potent α2-adrenergic receptor antagonist.[44][46][47] This metabolite may be responsible for the increased noradrenergic and dopaminergic activity observed with buspirone in animals.[46][48] In addition, 1-PP may play an important role in the antidepressant effects of buspirone.[48] Buspirone also has very weak and probably clinically unimportant affinity for the α1-adrenergic receptor.[35][49] However, buspirone has been reported to have shown “significant and selective intrinsic efficacy” at the α1-adrenergic receptor expressed in a “tissue- and species-dependent manner”.[49]
Unlike benzodiazepines, buspirone does not interact with the GABAA receptor complex.[2][50]
Pharmacokinetics
Buspirone has a low oralbioavailability of 3.9% relative to intravenous injection due to extensive first-pass metabolism.[2] The time to peak plasma levels following ingestion is 0.9 to 1.5 hours.[2] It is reported to have an elimination half-life of 2.8 hours,[2] although a review of 14 studies found that the mean terminal half-life ranged between 2 and 11 hours, and one study even reported a terminal half-life of 33 hours.[4] Buspirone is metabolized primarily by CYP3A4, and prominent drug interactions with inhibitors and inducers of this enzyme have been observed.[7][8] Major metabolites of buspirone include 5-hydroxybuspirone, 6-hydroxybuspirone, 8-hydroxybuspirone, and 1-PP.[4][5][6] 6-Hydroxybuspirone has been identified as the predominant hepatic metabolite of buspirone, with plasma levels that are 40-fold greater than those of buspirone after oral administration of buspirone to humans.[5] The metabolite is a high-affinity partial agonist of the 5-HT1A receptor (Ki = 25 nM) similarly to buspirone, and has demonstrated occupancy of the 5-HT1A receptor in vivo.[5] As such, it is likely to play an important role in the therapeutic effects of buspirone.[5] 1-PP has also been found to circulate at higher levels than those of buspirone itself and may similarly play a significant role in the clinical effects of buspirone.[46][48]
Buspirone was first synthesized, by a team at Mead Johnson, in 1968,[21] but was not patented until 1975.[54][55] It was initially developed as an antipsychoticdrug acting on the D2 receptor, but was found to be ineffective in the treatment of psychosis; it was then used as an anxiolytic instead.[2] In 1986, Bristol-Myers Squibb gained FDA approval for buspirone in the treatment of GAD.[21][56] The patent placed on buspirone expired in 2001 and it is now available as a generic drug.
Buspirone was primarily sold under the brand name Buspar.[57][59] Buspar is currently listed as discontinued by the US Federal Drug Administration.[60] In 2010, in response to a citizen petition, the US FDA determined that Buspar was not withdrawn for sale because of reasons of safety or effectiveness.[61]
Alkylation of 1-(2-pyrimidyl)piperazine (1) with 3-chloro-1-cyanopropane (2, 4-chlorobutyronitrile) gives 3, which is reduced either by hydrogenation over Raney nickel catalyst, or with LAH. The resulting 1° amine (4) from the previous step is then reacted with 3,3-tetramethyleneglutaric anhydride (5, 8-Oxaspiro[4.5]decane-7,9-dione) in order to yield buspirone (6).
A continuous flow method for the direct conversion of alcohols to amines via a hydrogen borrowing approach is reported. The method utilises a low loading (0.5%) of a commercial catalyst system ([Ru(p-cymene)Cl2]2 and DPEPhos), reagent grade solvent and is selective for primary alcohols. Successful methylation of amines using methanol and the direct dimethylamination of alcohols using commercial dimethylamine solution are reported. The synthesis of two pharmaceutical agents Piribedil (5) and Buspirone (25) were accomplished in good yields employing these new methods.
http://www.rsc.org/suppdata/c8/gc/c8gc03328e/c8gc03328e2.pdf 8-(4-hydroxybutyl)-8-azaspiro[4.5]decane-7,9-dione (23): A solution of 3,3-tetramethyleneglutaric anhydride (0.25 mol/L in THF) was combined in a tee piece with a solution of 4-amino-1-butanol (0.25 mol/L in THF) and reacted in a 20 mL reactor coil (stainless steel, 20 min residence time) heated at 250 °C. The output was concentrated in vacuo and the residue purified by column chromatography on silica gel to afford the product in 84% yield (Rf = 0.31, 63% DCM/AcOEt). 1H NMR (400 MHz, CDCl3) δ = 3.78 (t, J = 7.2 Hz, 2H), 3.65 (t, J = 6.0 Hz, 2H), 2.58 (s, 4H), 1.77 – 1.64 (m, 4H), 1.64 – 1.53 (m, 4H), 1.53 – 1.43 (m, 4H). 13C NMR (100 MHz, CDCl3) δ = 172.33, 62.28, 44.87, 39.47, 39.14, 37.54, 29.81, 24.35, 24.17. HRMS for [C13H22NO3] + calculated 240.1594 found 240.1605.
8-(4-(4-(pyrimidin-2-yl)piperazin-1-yl)butyl)-8-azaspiro[4.5]decane-7,9-dione (Buspirone, 25): The flow system was flushed with THF, the back-pressure regulator was set to 50 bar, and the coil reactor heated to 250 °C. Then a solution (10 mL overall volume) containing 1-(2-pyrimidyl)piperazine (2 mmol), 8-(4-hydroxybutyl)- 8-azaspiro[4.5]decane-7,9-dione (23) (2 mmol), dichloro(p-cymene)ruthenium(II) dimer (0.08 mmol) and bis[(2- diphenylphosphino)phenyl] ether (DPEPhos, 0.17 mmol) was pumped at 0.8 ml/min through a heated coil (8 mL, Phoenix reactor). The output solution obtained in steady state (monitored using the FlowUV) was concentrated in vacuo and purified by column chromatography on silica gel to afford the desired product in 76% yield (Rf = 0.29, 5% MeOH/DCM). 1H NMR (400 MHz, CDCl3) δ = 8.31 (d, J = 4.7 Hz, 2H), 6.48 (t, J = 4.7 Hz, 1H), 3.84 (t, J = 5.1 Hz, 4H), 3.79 (t, J = 6.8 Hz, 2H), 2.60 (s, 4H), 2.50 (t, J = 5.1 Hz, 4H), 2.40 (t, J = 6.8 Hz, 2H), 1.79 – 1.65 (m, 4H), 1.65 – 1.42 (m, 8H). 13C NMR (100 MHz, CDCl3) δ = 172.19, 161.63, 157.68, 109.77, 58.31, 53.06, 44.92, 43.60, 39.48, 39.35, 37.56, 26.04, 24.19, 24.19. HRMS for [C21H32N5O2] + calculated 386.2551 found 386.2570.
PAPER
Organic Preparations and Procedures International, 40(4), 391-394; 2008
The condensation of 1-(2-pyrimidinyl)piperazine (I) with 3-chloro-1-cyanopropane (II) by means of Na2CO3 in n-butanol gives 4-(2-pyrimidinyl)-1-(3-cyanopropyl)piperazine (III). This product is reduced with LiAlH4 or with H2 and Raney-Ni yielding 4-(2-pyrimidinyl)-1-(4-aminobutyl)piperazine (IV), which is finally condensed with 8-oxaspiro[4.5]decane-7,9-dione-(3,3-tetramethylene-glutaric anhydride) (V) in pyridine.
Buspirone, 8-[4-[4-(2-pyrimidyl)-1-piperazinyl]butyl]-8-azaspiro [4,5] decan-7,9-dione (5.2.6), is synthesized by the reaction of 1-(2-pyrimidyl)-4-(4-aminobutyl)piperazine (5.2.4) with 8-oxaspiro[4,5]decan-7,9-dione (5.2.5). In turn, 1-(2-pyrimidyl)-4-(4-aminobutyl)piperazine (5.2.4) is synthesized by the reaction of 1-(2-pyrimidyl)piperazine with 4-chlorobutyronitrile, giving 4-(2-pyrimidyl)-1-(3-cyanopropyl)piperazine (5.2.3), which is hydrogenated with Raney nickel into buspirone (5.2.4) [51–55].
Buspirone is an extremely specific drug that could possibly represent a new chemical class of anxiolytics—azaspirones. As an anxiolytic, its activity is equal to that of benzodiazepines; however, it is devoid of anticonvulsant and muscle relaxant properties, which are characteristic of benzodiazepines. It does not cause dependence or addiction. The mechanism of its action is not conclusively known. It does not act on the GABA receptors, which occurs in benzodiazepine use; however, it has a high affinity for seratonin (5-HT) receptors and a moderate affinity for dopamine (D2) receptors. Buspirone is effective as an anxiolytic. A few side effects of buspirone include dizziness, drowsiness, headaches, nervousness, fatigue, and weakness. This drug is intended for treatment of conditions of anxiety in which stress, muscle pain, rapid heart rate, dizziness, fear, etc. are observed; in other words, conditions of anxiety not associated with somewhat common, usual, and everyday stress. Synonyms for buspirone are anizal, axoren, buspar, buspimen, buspinol, narol, travin, and others.
Buspirone (Buspar®, 59, Figure 11.17) is a drug used for the treatment of anxiety and depression, thought to produce its effects by binding to the serotonin 5HT1A receptor [114–116]. Mainly as a result of hydroxylation reactions, it is extensively converted to various metabolites and blood concentrations return to low levels a few hours after dosing [117]. A major metabolite, 6-hydroxybuspirone, produced by the action of liver cytochrome P450 CYP3A4, was present at much higher concentrations in human blood than buspirone itself. For development of 6-hydroxybuspirone as a potential antianxiety drug, preparation and testing of the two enantiomers as well as the racemate was of interest. An enantioselective microbial reduction process was developed for the reduction of 6-oxobuspirone 60 to (R)-6-hydroxybuspirone 61a or (S)-6-hydroxybuspitone 61b. About 150 microbial cultures were screened for the enantioselective reduction of 60. Rhizopus stolonifer SC 13898, Neurospora crassa SC 13816, Mucor racemosus SC 16198, and Pseudomonas putida SC 13817 gave >50% reaction yields and >95% ee of (S)-6-hydroxybuspirone 61a. The yeast strains Hansenula polymorpha SC 13845 and Candida maltosa SC 16112 gave (R)-6-hydroxybuspirone in >60% reaction yield and >97% ee [118]. The NADPH-dependent (R)-reductase (RHBR) from H. polymorpha SC 13845 was purified to homogeneity, its N-terminal and internal amino acid sequences were determined and the corresponding gene was cloned and expressed in E. coli. To regenerate the NADPH required for reduction, glucose-6-phosphate dehydrogenase gene from Saccharomyces cerevisiae was cloned and coexpressed in the same E. coli strain. Recombinant cultures coexpressing (R)-reductase (RHBR) and glucose 6-phosphate dehydrogenase catalyzed the reduction of 6-ketobuspirone to (R)-6-hydroxybuspirone 61a in 99% yield and 99.9% ee at 50 g/L substrate input [119].
The NADH-dependent (S)-reductase (SHBR) from P. putida SC 16269 was also purified to homogeneity, its N-terminal and internal amino acid sequences were determined and the corresponding gene was cloned and expressed in E. coli. To regenerate the NADH required for reduction, the NAD+ dependent formate dehydrogenase gene from Pichia pastoris was also cloned and co-expressed in the same E. coli strain. Recombinant E. coli coexpressing (S)-reductase and formate dehydrogenase was used to catalyze the reduction of 6-ketobuspirone to (S)-6-hydroxybuspirone 61b, in >98% yield and >99.8% ee at 50 g/L substrate input [119].
The present invention relates to methods of treating anxiety and depression using R-6-hydroxy-buspirone and pharmaceutical compositions containing R-6-hydroxy-buspirone.
Buspirone, chemically: 8-[4-[4-(2-pyrimidinyl)1-piperazinyl]butyl-8-azaspiro(4,5)-decane-7,9-dione, is approved for the treatment of anxiety disorders and depression by the United States Food and Drug Administration. It is available under the trade name BUSPAR® from Bristol-Myers Squibb Company.
Studies have shown that buspirone is extensively metabolized in the body. (See, for example, Mayol, et al., Clin. Pharmacol. Ther., 37, p. 210, 1985). One of the metabolites is 6-hydroxy-8-[4-[4-(2-pyrimidinyl)1-piperazinyl]butyl-8-azaspiro(4,5)-decane-7,9-dione having Formula I. This metabolite is also known as BMS 28674, BMS 442608, or
as 6-hydroxy-buspirone. This compound is believed to be the active metabolite of buspirone and its use in treating anxiety disorders and depression is disclosed in U.S. Pat. No. 6,150,365. The specific stereochemistry of 6-hydroxy-buspirone has not been described previously. Neither racemic 6-hydroxy-buspirone nor its enantiomers are commercially available at the present time.
Preclinical studies demonstrate that 6-hydroxy-buspirone, like buspirone, demonstrates a strong affinity for the human 5-HT1A receptor. In functional testing, 6-hydroxy-buspirone produced a dose-dependent anxiolytic response in the rat pup ultrasonic vocalization test, a sensitive method for assessment of anxiolytic and anxiogenic effects (Winslow and Insel, 1991, Psychopharmacology, 105:513-520).
Clinical studies in volunteers orally dosed with buspirone demonstrate that 6-hydroxy-buspirone blood plasma levels were not only 30 to 40 times higher but were sustained compared to buspirone blood plasma levels. The time course of 6-hydroxy-buspirone blood plasma levels, unlike buspirone blood plasma levels, correlate more closely with the sustained anxiolytic effect seen following once or twice a day oral dosing with buspirone.
Although buspirone is an effective treatment for anxiety disorders and depression symptomatology in a significant number of patients treated, about a third of patients get little to no relief from their anxiety and responders often require a week or more of buspirone treatment before experiencing relief from their anxiety symptomatology. Further, certain adverse effects are reported across the patient population. The most commonly observed adverse effects associated with the use of buspirone include dizziness, nausea, headache, nervousness, lightheadedness, and excitement. Also, since buspirone can bind to central dopamine receptors, concern has been raised about its potential to cause unwanted changes in dopamine-mediated neurological functions and a syndrome of restlessness, appearing shortly after initiation of oral buspirone treatment, has been reported in small numbers of patients. While buspirone lacks the prominent sedative effects seen in more typical anxiolytics such as the benzodiazepines, patients are nonetheless advised against operating potentially dangerous machinery until they experience how they are affected by buspirone.
It can be seen that it is desirable to find a medicament with buspirone’s advantages but which demonstrates more robust anxiolytic potency with a lack of the above described adverse effects.
Formation of 6-hydroxy-buspirone occurs in the liver by action of enzymes of the P450 system, specifically CYP3A4. Many substances such as grapefruit juice and certain other drugs; e.g. erythromycin, ketoconazole, cimetidine, etc., are inhibitors of the CYP3A4 isozyme and may interfere with the formation of this active metabolite from buspirone. For this reason it would be desirable to find a compound with the advantages of buspirone but without the drug—drug interactions when coadministered with agents affecting the activity level of the CYP3A4 isozyme.
EXAMPLE 3One-Step Synthesis of 6-Hydroxy-buspirone (I)
Buspirone (19.3 g, 50 mmole) was dissolved in dry THF (400 mL) and the resulting solution was cooled to −78° C. A solution of KN(SiMe3)2 in toluene (100 mL, 1 M) was added slowly. After the reaction mixture was stirred at −78° C. for 1 h, a solution of 2-(phenylsulfonyl)-3-phenyloxaziridine (Davis reagent, prepared according to literature method: F. A. Davis, et al., Org. Synth., 1988, 66, 203) (17.0 g, 65 mmole) in dry THF (150 mL, precooled to −78° C.) was added quickly via a cannular. After stirred for 30 mins at −78° C., the reaction was quenched with 1 N HCl solution (500 mL). It was extracted with EtOAc (3×500 mL). The aqueous layer was separated, neutralized with saturated sodium bicarbonate solution, and extracted with EtOAc (3×500 mL). The combined organic extracts were dried over Na2SO4, filtered, and concentrated under reduced pressure to give a white solid residue which was subjected to column chromatography using CH2Cl2/MeOH/NH4OH (200:10:1) as the eluent to give pure 6-hydroxy-buspirone (I, 7.2 g) and a mixture of buspirone and 6-hydroxy-buspirone (I). The mixture was purified by above column chromatography to afford another 3.3 g of pure 6-hydroxy-buspirone (I).
^ Jump up to:ab Wong H, Dockens RC, Pajor L, Yeola S, Grace JE, Stark AD, et al. (August 2007). “6-Hydroxybuspirone is a major active metabolite of buspirone: assessment of pharmacokinetics and 5-hydroxytryptamine1A receptor occupancy in rats”. Drug Metabolism and Disposition. 35 (8): 1387–92. doi:10.1124/dmd.107.015768. PMID17494642. S2CID25558546.
^ Jump up to:abcd Zhu M, Zhao W, Jimenez H, Zhang D, Yeola S, Dai R, et al. (April 2005). “Cytochrome P450 3A-mediated metabolism of buspirone in human liver microsomes”. Drug Metabolism and Disposition. 33 (4): 500–7. doi:10.1124/dmd.104.000836. PMID15640381. S2CID10142905.
^ Sontheimer DL, Ables AZ (March 2001). “Is imipramine or buspirone treatment effective in patients wishing to discontinue long-term benzodiazepine use?”. The Journal of Family Practice. 50(3): 203. PMID11252203.
^ Prisco V, Iannaccone T, Di Grezia G (2017-04-01). “Use of buspirone in selective serotonin reuptake inhibitor-induced sleep bruxism”. European Psychiatry. Abstract of the 25th European Congress of Psychiatry. 41: S855. doi:10.1016/j.eurpsy.2017.01.1701.
^ Lamberg TS, Kivistö KT, Laitila J, Mårtensson K, Neuvonen PJ (1998). “The effect of fluvoxamine on the pharmacokinetics and pharmacodynamics of buspirone”. European Journal of Clinical Pharmacology. 54 (9–10): 761–6. doi:10.1007/s002280050548. PMID9923581. S2CID21939719.
^ Jump up to:abc Roth BL, Driscol J. “PDSP Ki Database”. Psychoactive Drug Screening Program (PDSP). University of North Carolina at Chapel Hill and the United States National Institute of Mental Health. Retrieved 14 August 2017.
^ Nelson DR, Thomas DR (May 1989). “[3H]-BRL 43694 (Granisetron), a specific ligand for 5-HT3 binding sites in rat brain cortical membranes”. Biochemical Pharmacology. 38 (10): 1693–5. doi:10.1016/0006-2952(89)90319-5. PMID2543418.
^ Jump up to:ab Borsini F, Giraldo E, Monferini E, Antonini G, Parenti M, Bietti G, Donetti A (September 1995). “BIMT 17, a 5-HT2A receptor antagonist and 5-HT1A receptor full agonist in rat cerebral cortex”. Naunyn-Schmiedeberg’s Archives of Pharmacology. 352 (3): 276–82. doi:10.1007/bf00168557. PMID8584042. S2CID19340842.
^ Plassat JL, Amlaiky N, Hen R (August 1993). “Molecular cloning of a mammalian serotonin receptor that activates adenylate cyclase”. Molecular Pharmacology. 44 (2): 229–36. PMID8394987.
^ Lovenberg TW, Baron BM, de Lecea L, Miller JD, Prosser RA, Rea MA, et al. (September 1993). “A novel adenylyl cyclase-activating serotonin receptor (5-HT7) implicated in the regulation of mammalian circadian rhythms”. Neuron. 11 (3): 449–58. doi:10.1016/0896-6273(93)90149-l. PMID8398139. S2CID28729004.
^ Jump up to:ab Blier P, Curet O, Chaput Y, de Montigny C (July 1991). “Tandospirone and its metabolite, 1-(2-pyrimidinyl)-piperazine–II. Effects of acute administration of 1-PP and long-term administration of tandospirone on noradrenergic neurotransmission”. Neuropharmacology. 30 (7): 691–701. doi:10.1016/0028-3908(91)90176-c. PMID1681447. S2CID44297577.
^ Zuideveld KP, Rusiç-Pavletiç J, Maas HJ, Peletier LA, Van der Graaf PH, Danhof M (December 2002). “Pharmacokinetic-pharmacodynamic modeling of buspirone and its metabolite 1-(2-pyrimidinyl)-piperazine in rats”. The Journal of Pharmacology and Experimental Therapeutics. 303 (3): 1130–7. doi:10.1124/jpet.102.036798. PMID12438536. S2CID14139919.
^ Dockens RC, Salazar DE, Fulmor IE, Wehling M, Arnold ME, Croop R (November 2006). “Pharmacokinetics of a newly identified active metabolite of buspirone after administration of buspirone over its therapeutic dose range”. Journal of Clinical Pharmacology. 46(11): 1308–12. doi:10.1177/0091270006292250. PMID17050795.
^ Jajoo HK, Mayol RF, LaBudde JA, Blair IA (1989). “Metabolism of the antianxiety drug buspirone in human subjects”. Drug Metabolism and Disposition. 17 (6): 634–40. PMID2575499.
Pyridostigmine BromideCAS Registry Number: 101-26-8CAS Name: 3-[[(Dimethylamino)carbonyl]oxy]-1-methylpyridinium bromideAdditional Names: 3-hydroxy-1-methylpyridinium bromide dimethylcarbamate; 1-methyl-3-hydroxypyridinium bromide dimethylcarbamate; 3-(dimethylcarbamyloxy)-1-methylpyridinium bromideManufacturers’ Codes: Ro-1-5130Trademarks: Kalymin (Temmler); Mestinon (Roche); Regonol (Organon)Molecular Formula: C9H13BrN2O2Molecular Weight: 261.12Percent Composition: C 41.40%, H 5.02%, Br 30.60%, N 10.73%, O 12.25%Literature References: Reversible inhibitor of acetylcholinesterase. Prepn: Urban, US2572579 (1951 to Hoffmann-La Roche). Mechanism of protective effect in soman poisoning: X. Deyi et al.,Fundam. Appl. Toxicol.1, 217 (1981). Evaluation of effect on neuromuscular function: M. Glikson et al.,ibid.16, 288 (1991). Evaluation of side effects profile under desert conditions: J. E. Cook et al.,Mil. Med.157, 250 (1992). Review of prophylactic effect in nerve agent poisoning: R. M. Dawson, J. Appl. Toxicol.14, 317 (1994).Properties: Shiny, hygroscopic crystals from abs ethanol, mp 152-154°. Freely sol in water, alcohol. Practically insol in ether, acetone, benzene. Aq solns may be sterilized by autoclaving with steam.Melting point: mp 152-154°Therap-Cat: Cholinergic; in treatment of myasthenia gravis. Pre-exposure antidote to chemical warfare agents.Keywords: Cholinergic.
Pyridostigmine is used to treat muscle weakness in people with myasthenia gravis or forms of congenital myasthenic syndrome and to combat the effects of curariform drug toxicity. Pyridostigmine bromide has been FDA approved for military use during combat situations as an agent to be given prior to exposure to the nerve agent Soman in order to increase survival. Used in particular during the first Gulf War, pyridostigmine bromide has been implicated as a causal factor in Gulf War syndrome.[5]
Pyridostigmine sometimes is used to treat orthostatic hypotension.[6] It may also be of benefit in chronic axonal polyneuropathy.[7]
Pyridostigmine bromide is contraindicated in cases of mechanical intestinal or urinary obstruction and should be used with caution in patients with bronchial asthma.[9][10]
Pyridostigmine inhibits acetylcholinesterase in the synaptic cleft, thus slowing down the hydrolysis of acetylcholine. It is a quaternary carbamate inhibitor of cholinesterase that does not cross the blood–brain barrier which carbamylates about 30% of peripheral cholinesterase enzyme. The carbamylated enzyme eventually regenerates by natural hydrolysis and excess ACh levels revert to normal.
The ACh diffuses across the synaptic cleft and binds to receptors on the post synaptic membrane, causing an influx of Na+, resulting in depolarization. If large enough, this depolarization results in an action potential. To prevent constant stimulation once the ACh is released, an enzyme called acetylcholinesterase is present in the endplate membrane close to the receptors on the post synaptic membrane, and quickly hydrolyses ACh.
Names
Pyridostigmine bromide is available under the trade names Mestinon (Valeant Pharmaceuticals), Regonol and Gravitor (SUN Pharma).
Chemistry
Pyridostigmine, 3-[(dimethylaminocarbonyl)oxy]-1-methyl pyridinium bromide, is synthesized from 3-hydroxypyridine, which is reacted with dimethylaminocarbamoyl chloride, which gives 3-(dimethylaminocarbamoyl)pyridine. The last is reacted with methylbromide, giving pyridostigmine.
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
^ Gales BJ, Gales MA (2007). “Pyridostigmine in the treatment of orthostatic intolerance”. Annals of Pharmacotherapy. 41 (2): 314–8. doi:10.1345/aph.1H458. PMID17284509. S2CID22855759.
^ Kanjwal K, Karabin B, Sheikh M, et al. (June 2011). “Pyridostigmine in the treatment of postural orthostatic tachycardia: a single-center experience”. Pacing and Clinical Electrophysiology. 34 (6): 750–5. doi:10.1111/j.1540-8159.2011.03047.x. PMID21410722. S2CID20405336.
ORD +41.3 °, water, 4% ; Wavlen: 589.3 nm; Temp: 25 °C, AND MP 163-165 °C, GB 747768 1956 NorepinephrineCAS Registry Number: 51-41-2CAS Name: 4-[(1R)-2-Amino-1-hydroxyethyl]-1,2-benzenediolAdditional Names: (-)-a-(aminomethyl)-3,4-dihydroxybenzyl alcohol; l-3,4-dihydroxyphenylethanolamine; noradrenaline; levarterenolTrademarks: Adrenor; Levophed (Winthrop)Molecular Formula: C8H11NO3Molecular Weight: 169.18Percent Composition: C 56.79%, H 6.55%, N 8.28%, O 28.37%Literature References: Demethylated precursor of epinephrine, q.v. Occurs in animals and man, and is a sympathomimetic hormone of both adrenal origin and adrenergic orthosympathetic postganglionic origin in man. Physiologic review: Malmejac, Physiol. Rev.44, 186 (1964). It has also been found in plants, e.g., Portulaca olerocea L., Portulacaceae: Fing et al.,Nature191, 1108 (1961). Synthesis of dl-form: Payne, Ind. Chem.37, 523 (1961). Historic review of synthesis: Loewe, Arzneim.-Forsch.4, 583 (1954). Resolution of dl-form: Tullar, J. Am. Chem. Soc.70, 2067 (1948); idem,US2774789 (1956 to Sterling Drug). Configuration: Pratesi et al.,J. Chem. Soc.1959, 4062. Comprehensive description: C. F. Schwender, Anal. Profiles Drug Subs.1, 149-173 (1972); T. D. Wilson, ibid.11, 555-586 (1982).Properties: Microcrystals, dec 216.5-218°. [a]D25 -37.3° (c = 5 in water with 1 equiv HCl).Optical Rotation: [a]D25 -37.3° (c = 5 in water with 1 equiv HCl) Derivative Type: HydrochlorideCAS Registry Number: 329-56-6Trademarks: Arterenol (HMR)Molecular Formula: C8H11NO3.HClMolecular Weight: 205.64Percent Composition: C 46.73%, H 5.88%, N 6.81%, O 23.34%, Cl 17.24%Properties: Crystals, mp 145.2-146.4°. [a]D25 -40° (c = 6). Freely sol in water. Solns slowly oxidize under the influence of light and oxygen in a manner comparable to epinephrine hydrochloride.Melting point: mp 145.2-146.4°Optical Rotation: [a]D25 -40° (c = 6) Derivative Type:d-BitartrateCAS Registry Number: 69815-49-2Additional Names: Levarterenol bitartrateTrademarks: Aktamin; BinodrenalMolecular Formula: C8H11NO3.C4H6O6Molecular Weight: 319.26Percent Composition: C 45.14%, H 5.37%, N 4.39%, O 45.10%Properties: Obtained as the monohydrate, crystals, mp 102-104°. [a]D25 -10.7° (c = 1.6 in H2O). When anhydr, mp 158-159° (some decompn). Freely sol in water.Melting point: mp 102-104°; mp 158-159° (some decompn)Optical Rotation: [a]D25 -10.7° (c = 1.6 in H2O) Derivative Type:dl-FormProperties: Crystals, dec 191°. Sparingly sol in water; very slightly sol in alc, ether; readily sol in dilute acids, caustic. Therap-Cat: Adrenergic (vasopressor); antihypotensive.Therap-Cat-Vet: Sympathomimetic; vasopressor in shock.Keywords: a-Adrenergic Agonist; Antihypotensive.
Precursor of epinephrine that is secreted by the adrenal medulla and is a widespread central and autonomic neurotransmitter. Norepinephrine is the principal transmitter of most postganglionic sympathetic fibers and of the diffuse projection system in the brain arising from the locus ceruleus. It is also found in plants and is used pharmacologically as a sympathomimetic.
Norepinephrine (sometimes referred to as l-arterenol/Levarterenol or l-norepinephrine) is a sympathomimetic amine which differs from epinephrine by the absence of a methyl group on the nitrogenatom.
Norepinephrine Bitartrate is (-)-α-(aminomethyl)-3,4-dihydroxybenzyl alcohol tartrate (1:1) (salt) monohydrate and has the following structural formula:
LEVOPHED is supplied in sterile aqueous solution in the form of the bitartrate salt to be administered by intravenous infusion following dilution. Norepinephrine is sparingly soluble in water, very slightly soluble in alcohol and ether, and readily soluble in acids. Each mL contains the equivalent of 1 mg base of norepinephrine, sodium chloride for isotonicity, and not more than 2 mg of sodium metabisulfite as an antioxidant. It has a pH of 3 to 4.5. The air in the ampuls has been displaced by nitrogen gas.
Common side effects include headache, slow heart rate, and anxiety.[2] Other side effects include an irregular heartbeat.[2] If it leaks out of the vein at the site it is being given, norepinephrine can result in limb ischemia.[2] If leakage occurs the use of phentolamine in the area affected may improve outcomes.[2] Norepinephrine works by binding and activating alpha adrenergic receptors.[2]
Norepinephrine was discovered in 1946 and was approved for medical use in the United States in 1950.[2][4] It is available as a generic medication.[2]
Norepinephrine is the INN while noradrenaline is the BAN.
SYN
Chemical Synthesis
Norepinephrine, L-1-(3,4-dihydroxyphenyl)-2-aminoethanol (11.1.4), is synthesized by two methods starting from 3,4-dihydroxybenzaldehyde. According to the first method, the indicated aldehyde is transformed into the cyanohydrin (11.1.3) by reaction with hydrogen cyanide, which is then reduced into norepinephrine (11.1.5). The second method consists of the condensation of diacetate of the same aldehyde with nitromethane, which forms (3,4-diacetoxyphenyl)-2-nitroethanol (11.1.5). Then the nitro group is reduced and the product (11.1.6) is hydrolyzed into the desired norepinephrine (11.1.4) [4,9,13,14].
Purification Methods
Recrystallise adrenor from EtOH and store it in the dark under N2. [pKa, Lewis Brit J Pharmacol Chemother 9 488 1954, UV: Bergstr.m et al. Acta Physiol Scand 20 101 1950, Fluorescence: Bowman et al. Science NY 122 32 1955, Tullar J Am Chem Soc 70 2067 1948.] The L-tartrate salt monohydrate has m 102-104.5o, [] D -11o (c 1.6, H2O), after recrystallisation from H2O or EtOH. [Beilstein 13 III 2382.]
PATENT
https://patents.google.com/patent/WO2013008247A1/en4-[(lR)-2-amino-l-hydroxyethyl]benzene-l,2-diol, commonly known as (R)-(-)- norepinephrine or noradrenaline is a catecholamine with multiple roles including as a hormone and a neurotransmitter. As a stress hormone, norepinephrine affects parts of the brain where attention and responding actions are controlled. Along with epinephrine, norepinephrine also underlies the fight-or-flight response, directly increasing heart rate, triggering the release of glucose from energy stores, and increasing blood flow to skeletal muscle. Norepinephrine also has a neurotransmitter role when released diffusely in the brain as an antiinflammatory agent.When norepinephrine acts as a drug it increases blood pressure by increasing vascular tone through a-adrenergic receptor activation. The resulting increase in vascular resistance triggers a compensatory reflex that overcomes its direct stimulatory effects on the heart, called the baroreceptor reflex, which results in a drop in heart rate called reflex bradycardia.(R)-(-)-Norepinephrine has a following structure:
(R)-(-)-Norepinephrine was first time disclosed in the US patent US2774789, where it was obtained by resolution of dl-norepinephrine, with optically active acids such as d- tartaric acid, 1-malic acid or N-benzoyl-l-threonine. The patent does not disclose the preparation of dl-norepinephrine. The patent GB747768 describes reduction of amino ketones where 3,4-dihydroxy-a- aminoacetophenone hydrochloride was converted into its d-tartrate salt; followed by reduction of the d-tartrate salt. This process leads to formation of excessive amount of d- adrenaline d-tartrate (which is a bi-product) as it crystallized first; whereas the desired 1- adrenaline d-tartrate crystallizes after 2 days and in smaller yield. Also the patent does not disclose the source of 3,4-dihydroxy-a-aminoacetophenone hydrochloride.It has been unsuccessfully tried to treat dihydroxy-a-chloroacetophenone with hexamethylenetetramine (commonly known as hexamine) and to treat the reaction product with an acid to obtain arterenone (see Mannich, Hahn B., Berichte der deutschen chemischen Gesellschaft, volume 44, issue 2, Pages 1542 – 1552 (1911)). Mannich found that the treatment of this and similar halogen ketones with hexamine did not produce an addition compound but resulted in splitting of halogen acid which made the process impossible. Mannich also found that an addition compound of the halogen ketone and hexamine is formed only when the two phenolic hydroxyl groups are closed i.e. protected by acylation or etherification. Hence according to Mannich, the reaction is not at all possible for the compounds containing two unprotected phenolic hydroxyl groups. The US patent US 1680055 discloses the preparation of monohydroxy-a-substituted- aminoacetophenones either by reacting monohydroxy-a-bromoacetophenones with a substituted amine or by reacting protected monohydroxy-a-bromoacetophenones with a substituted amine followed by deprotection. The patent does not disclose the preparation of dihydroxy-a-aminoacetophenones (where amino group is unsubstituted).It is disclosed in the US patent US2786871 that when chloroaceto pyrocatechol is treated with ammonia, arterenone is obtained in 50% yield. However when the reaction is carried out in basic medium, darkening of the reaction mass takes place which results in coloured product. The patent also discloses preparation of amino-methyl-(monohydroxyphenyl)- ketones by reacting halogen ketone with hexamine. It is also disclosed in the patent that the process is applicable only to the halogenomethyl-monohydroxyphenyl-ketones.Following are some of the methods for preparation of 3,4-dihydroxy-a- aminoacetophenone, reported in the literature. J. Am. Pharm. Association (1946) 35, 306 – 309 discloses preparation of 3,4-dihydroxy- a-aminoacetophenone by reacting 3,4-dihydroxy-a-chloroacetophenone with dibenzyl amine followed by hydrogenation of resulting dibenzylamino ketone. The main disadvantage of this reaction is formation of derivatives of dibenzyl amines, which remain in the final product in the form of impurities.Acta Chimica Academiae Scientiarum Hungaricae (1951), 1, 395-402, discloses preparation of 3,4-dihydroxy-a-aminoacetophenone from 3,4-dihydroxyphenyloxo acetaldehyde and benzyl amine followed by reduction of benzylamino ketone intermediate. The main disadvantage of this method is that the starting acetaldehyde derivative is very expensive and not easily available.It is disclosed in Recueil des Travaux Chimiques des Pays-Bas et de la Belgique (1952), 71, 933-44, that 3,4-dihydroxy-a-aminoacetophenone hydrochloride is formed by demethylation of 3,4-dimethoxy-a-aminoacetophenone hydrochloride using 48% HBr. The reaction results in less than 10% yield of the aminoacetophenone.Monatshefte fuer Chemie (1953), 84 1021-32, discloses preparation of 3,4-dihydroxy-a- aminoacetophenone by reacting 3,4-dihydroxy-a-chloroacetophenone with sodium azide followed by hydrogenation of azide intermediate using 4% palladium on carbon as a catalyst. In the hydrogenation step, 1.6 gm of azide intermediate requires 1.4 gm of catalyst, which is not economical and industrially feasible.
Preparation of 3,4-dihydroxy-a-aminoacetophenones hydrochloride is disclosed in J. Am. Chem. Soc, 1955, volume 77, issue 10, pages 2896 – 2897. The following scheme is disclosed in the article:
It is clear from the above scheme that the process requires additional steps of protection and deprotection of hydroxyl and amino groups, and use of potassium phthalimide requires anhydrous reaction conditions. Therefore the process is time consuming and not economical.Chinese patent CN101798271A describes reduction of 3,4-dihydroxy-a- aminoacetophenone hydrochloride in water as solvent followed by neutralization with aqueous ammonia. Since dl-norepinephrine has partial solubility in aqueous basic medium result in to loss of product. Also it is necessary to maintain low volume of solvent throughout the process for better yields making the process stringent.European patent EP1930313 discloses preparation of a-amino ketones. The preparation is carried out by reacting an organic sulfide in a polar solvent with a compound containing a leaving group attached to a primary or secondary carbon atom to form a sulfonium salt, which is reacted with a ketone in presence of a base and a polar solvent. Oxiranes obtained are further converted into the corresponding aminoketone, by aminolysis followed by selective oxidation. The following scheme is disclosed in the patent.
It is clear from the above scheme that the process requires many steps and hence is time consuming. The patent does not exemplify the synthesis of dihydroxy-a- aminoacetophenones.Thus, the search for a suitable manufacturing process for (R)-norepinephrine intermediates remains undoubtedly of interest. We were surprised to find that hardly any literature discloses the process for preparation of dihydroxy-a-aminoacetophenones acid addition salts. We have found that the reaction of dihydroxy-a-haloacetophenone with hexamine is feasible and results in high yield of product although both the hydroxyl groups on the phenyl ring of acetophenone are unprotected. Object of the invention:It is therefore an object of the invention is to overcome or ameliorate at least one disadvantage of the prior art or to provide a useful alternative.Another object of the invention is to provide a novel, safe, efficient, concise, ecological, high yielding, industrially feasible and simpler process for preparation of (R)-(-)- norepinephrine intermediates.Another object of the invention is to provide a process for synthesis of 3,4-dihydroxy-a- aminoacetophenone salt, which is feasible without protecting both the hydroxyl group on the phenyl ring of acetophenone.Yet another object of the invention is to provide an improved process for hydrogenation of 3,4-dihydroxy-a-aminoacetophenone salt to prepare (dl)-norepinephrine salt.Summary of the invention:In accordance with the above objectives, the present invention provides a process for preparation of (dl)-norepinephrine intermediate of formula (III) comprising reacting 3,4- dihydroxy-a-haloacetophenone of formula (I) with hexamine to provide a quaternary ammonium salt of formula (II); followed by hydrolyzing the quaternary ammonium salt of formula (II) with an acid.In a second aspect, the present invention provides a novel quaternary ammonium salt of formula (II) and its preparation.In a third aspect, the present invention provides a novel process for hydrogenation of 3,4- dihydroxy-a-aminoacetophenone acid salt to provide (dl)-norepinephrine acid addition salt.Example 1Preparation of quaternary ammonium saltA 5000 ml four neck round bottom flask with water condenser and calcium chloride tube was charged with Hexamine (210.28 gm), chloroform (1200 ml), 3,4-dihydroxy-a- chloroacetophenone (250 gm) and isopropanol (1000 ml) at room temperature. The reaction mass was gently heated at 63°C for 4 hours. The reaction was monitored by TLC. The reaction mass was cooled to room temperature and filtered to get solid. The solid was washed with acetone and dried at 50°C for 4 hours to obtain quaternary ammonium salt which was used in the next step without purification.Yield – 410 gm (93.65%)Nature – off white solidm.p. – 180 to l82°CNMR (DMSO-d6): – δ =4.51 – 4.75 (m, 8H), 5.39 (s, 6H), 6.92 (d, 1H, J= 7.5 Hz), 7.37 – 7.42 (m, 2H), 9.67 (s, br, 1H), 10.44 (s, br, 1H)Example 2Preparation of 3,4-dihydroxy-a-aminoacetophenone hydrochlorideA 2000 ml four neck round bottom flask with water condenser and calcium chloride tube was charged with the quaternary ammonium salt obtained in the example 1 (120 gm), methanol (862.5 ml) and cone, hydrochloric acid (194.4 ml). The reaction mixture was heated to 60 to 65°C and aged at same temperature for 3 to 4 hours. The reaction was monitored by TLC. The reaction mass was cooled and neutralized using base to give 3,4- dihydroxy-a-aminoacetophenone. The solid was filtered, washed with water and dried at 50°C. This base was further converted in to its hydrochloride salt with IPA-HC1 mixture. Yield – 72 gm (96.3%)Nature – off white solidHPLC – 99.7%1H NMR(CD30D) – 5 = 3.62(s, 1H), 6.80 (d, J = 8 Hz, 1H), 7.38 (d, J = 1.3 Hz, 1H), 7.63 (d, J = 8 Hz, 1H).Example 3Preparation of (dl)-norepinephrine hydrochlorideA 500 ml hydrogenation flask was charged with 3,4-dihydroxy-a-aminoacetophenone hydrochloride obtained in the example 2 (55 gm), 10% palladium on carbon (5 gm) and methanol (300 ml). The reaction mixture was heated to 45°C with hydrogen gas pressure of 4 to 5 kg m2. The reaction mixture was stirred at 45°C for 5 hours. The catalyst was removed by filtration. The filtrate was cooled to 5 to 10 °C and ammonia gas was passed through the solvent for 2 h till the pH of the solution was around 9. The solid obtained was filtered, washed with methanol and dried in air to obtain (dl)-norepinephrine. Yield – 43.5 gm (96.7%)Nature white crystalline solidHPLC 99.6%Example 4Preparation of (dl)-norepinephrine hydrochlorideA 500 ml hydrogenation flask was charged with 3,4-dihydroxy-a-aminoacetophenone hydrochloride obtained from process similar to example 2 (55 gm), 10% palladium on carbon (5 gm) and methanol (300 ml). The reaction mixture was aged at 25 °C with hydrogen gas pressure of 4 to 3 kg/m2. The reaction mixture was stirred at 25°C for 15 hours. The reaction was monitored by TLC. The catalyst was removed by filtration. The filtrate was cooled to 5 to 10 °C and ammonia solution was added to the reaction mixture till the pH of the solution around 9. The solid obtained was filtered, washed with methanol and dried in air to obtain (dl)-norepinephrine.Yield – 41.5 gm (92.2%)Nature – white crystalline solidHPLC – 99.5% PATENTUS-10865180https://patentscope.wipo.int/search/en/detail.jsf?docId=US283323778&_cid=P11-KMEC1N-93277-1
Norepinephrine Bitartrate (Arterenol Bitartrate) is chemically known as (−)-α-(aminomethyl)-3, 4-dihydroxybenzyl alcohol tartrate (1:1) (salt) monohydrate is a catecholamine family that functions in the brain and body as a hormone and neurotransmitter. As a stress hormone, Norepinephrine affects parts of the brain where attention and responding actions are controlled. Along with epinephrine, Norepinephrine also underlies the fight-or-flight response, directly increasing heart rate, triggering the release of glucose from energy stores, and increasing blood flow to skeletal muscle. Norepinephrine also has a neurotransmitter role when released diffusely in the brain as an anti-inflammatory agent.
LEVOPHED® (l-Norepinephrine) is supplied in sterile aqueous solution in the form of the bitartrate salt to be administered by intravenous infusion following dilution. Norepinephrine is sparingly soluble in water, very slightly soluble in alcohol and ether, and readily soluble in acids. Each ml contains the equivalent of 1 mg base of Norepinephrine, sodium chloride for isotonicity, and not more than 2 mg of sodium metabisulfite as an antioxidant.
Norepinephrine Bitartrate is (−)-α-(amino methyl)-3,4-dihydroxybenzyl alcohol tartrate (1:1) (salt) monohydrate and has the following structural formula:
(l)-Norepinephrine was first disclosed in 1947 by Sterling Drugs. U.S. Pat. No. 2,774,789 discloses the resolution of dl-Norepinephrine with optically active acids such as d-tartaric acid, 1-malic acid or N-benzoyl-l-threonine. The patent does not disclose the basic synthesis of dl-Norepinephrine.
Journal of the American Chemical Society, Volume 70 (6), 1948 describes the resolution of dl-Norepinephrine in to d-arterenol-d-bitartrate and l-arterenol-d-bitartrate in water and aqueous methanol. Further it also describes isolation of d-arterenol and l-arterenol form above tartrate salts.
U.S. Pat. No. 2,786,871 discloses the process for the preparation of arterenol wherein chloroacetopyrocatechol is treated with ammonia and arterenol is obtained in 50% yield.
J. Am. Pharm. Association (1946) 35, 306-309 discloses preparation of 3,4-dihydroxyaminoacetophenone by reacting 3,4-dihydroxy-α-chloroacetophenone with dibenzyl amine, followed by hydrogenation of the resulting dibenzylamino ketone. The main disadvantage of this reaction is the formation of derivatives of dibenzyl amines, which carried over to final product in the form of impurities.
Acta Chimica Academiae Scientiarum Hungaricae (1951), 1, 395-402 discloses preparation of 3, 4-dihydroxy-α-aminoacetophenone from 3,4-dihydroxyphenyloxo acetaldehyde and benzyl amine followed by reduction of the benzylamino ketone intermediate. The main disadvantage of this method is that the starting acetaldehyde derivative is very expensive and not easily available.
CN101798271A describes reduction of 3,4-dihydroxy-α-aminoacetophenone hydrochloride in water as solvent followed by neutralization with aqueous ammonia. Since dl-Norepinephrine has partial solubility in aqueous basic medium, this process results in a loss of product. Also, it is necessary to maintain low volume of solvent throughout the process for better yields making the process stringent.
WO2009004593 describes the process for the preparation of Epinephrine wherein (−) epinephrine is obtained by chiral separation of dl-epinephrine using the chiral acid such as L-tartaric acid with an optical purity of 95.24%.
WO2013008247 discloses a process for preparation of (dl)-norepinephrine hydrochloride salt by reacting 3,4-dihydroxy-a-haloacetophenone with hexamethylenetetramine to provide hexamine salt; followed by hydrolysis and hydrogenation. However, this process fails to teach the resolution of (dl)-norepinephrine hydrochloride and preparation of l-Norepinephrine Bitartrate monohydrate.
WO2016038422 discloses a process for the preparation of optically enriched adrenaline or adrenaline tartrate comprising the steps of: (a) reacting a mixture of (−)-adrenaline and (+)-adrenaline with L(+)-tartaric acid to form adrenaline tartrate; (b) contacting the adrenaline tartrate with less than 1 equivalent of ammonium hydroxide. However, the product achieved is with purity of only 98%.
CN107298646 describes the process for the preparation of Norepinephrine wherein L-Norepinephrine tartrate is obtained by chiral separation of dl-Norepinephrine using the chiral acid such as L-tartaric acid. The chiral separation step using L-tartaric acid is repeated once to obtain pure Norepinephrine. However, there is no information on bitartrate salt and its optical purity.
In light of the above, there remains a need in the art for highly pure l-Norepinephrine Bitartrate having high enantiomeric purity i.e. greater than 99.0% so as to provide enhanced therapeutic efficacy and safety when administered. Surprisingly the present inventors have found out a process for the preparation of (l)-Norepinephrine Bitartrate having enantiomeric purity greater than 99.5%, for which protection is sought.
To a four necked 100 ml flask charged racemic Norepinephrine base (20 gm), d-(−) tartaric acid (18.34 gm), and water (35 ml) at room temperature. The reaction mass was stirred to obtain clear solution, cooled to 0-5° C. After 5 hours slight turbidity was observed. Turbidity increases slowly to get thick white slurry after 6 hours, reaction mass becomes very thick which was difficult to filter, washed solid wet cake by 4.0 ml water followed by two 12 ml portions of 95% ethanol. Suck dried the solid completely, dried at 45° C. to get l-Norepinephrine Bitartrate (28 gm) which is in crude form.
Crude l-Norepinephrine Bitartrate (20 gm) dissolved in 14 ml of water at 50° C. Clear solution was obtained. Activated charcoal was added to this solution and stirred the reaction mass for more for 30 min. Filtered through Hyflo and cooled to 0-5° C. After 2 hours, clear solution obtained gets converted to thick solid mass. Filtered and washed the solid with 1.5 ml of chilled water followed 14 ml of 95% ethanol.
This dry solid 8 gm (after 1 st purification) was then dissolved in 8 ml of water at 50° C. to get clear solution. This reaction mass was then cooled to 0-5° C. After 1 hour, a clear solution gets converted to a thick solid mass. Maintained the reaction mass for more than 2 hours at the same conditions. Filtered the thick solid and washed with 95% ethanol. Dried the solid at 45° C. to obtain l-Norepinephrine Bitartrate.
Chiral Purity by HPLC: l-Norepinephrine Bitartrate=68.45%, and d-isomer=31.55%
To a four necked flask charged racemic Norepinephrine base (20 gm), d-(−) tartaric acid (18.34 gm), and water (35.20 ml) at room temperature. After 5 minutes reaction mass becomes clear liquid. Cooled the reaction mass to 2-3° C. After 30 minutes, reaction mass was observed to be turbid and further the reaction mass becomes very thick. This mass was, stirred for 2 hours at 0-5° C. Then filtered reaction mass at same temperature and washed solid wet cake with 3.5 ml water followed by two 11.8 ml portions of 95% ethanol. Dried the solid at air oven at 45° C. to get crude tartrate salt (15 gm).
Crude tartrate salt (15 gm) was dissolved in 5 ml of water at 50° C. to get clear solution. Cooled to 2-3° C. After 30 minutes, a clear solution gets converted to a thick solid mass. Filtered the solid and washed with 1.5 ml of chilled water and then 15 ml of 95% ethanol. Dried the solid at 45° C. to obtain semi pure l-Norepinephrine Bitartrate (8 gm).
This semi pure l-Norepinephrine Bitartrate (8 gm) was dissolved in 8 ml of water at 50° C. to get clear solution. Cooled the mass to 2-3° C. After 30 minutes clear solution gets converted to thick solid mass. Filtered the solid and washed with 8 ml of 95% ethanol. Dried the solid at 45° C. to obtain pure l-Norepinephrine Bitartrate (3 gm).
Example-1: Preparation of 2-Chloro-1-(3, 4-Dihydroxyacetophenone)
In round bottom flask, charged Methylene Chloride (1000 ml), Aluminium chloride (300 gm) and cooled to 0-5° C. Pyrocatechol (100 gm) was added lot wise. Chloroacetyl chloride (108 gm) was added drop wise at 0-5° C. Then stirred the reaction mass at 25-30° C. for 20-24 hours. After completion of the reaction, reaction mass was quenched in aq. HCl, filtered the reaction mass and wet cake was charged in water containing acetic acid. Filtered the reaction mass and cooled to 15-20° C., filtered solid and washed with water.
Yield: 110 gm.
HPLC Purity: 99.5%
Example-2: Preparation of Hexamine Salt
In a round bottom flask charged 2-chloro-1-(3, 4-dihydroxyacetophenone) (100 gm), Hexamine (87 gm), IPA (500 ml), Chloroform (400 ml). Stirred the reaction mass at reflux temperature for 6 hours. After completion of the reaction, cooled to 25-30° C., filtered and washed the wet cake with IPA and Methanol.
Yield: 160 gm.
HPLC Purity: 99.3%
Example-3: Preparation of 2-Amino-1-(3,4-Dihydroxyphenyl)Ethanone Hydrochloride
In a round bottom flask charged Hexamine salt (100 gm), Methanol (600 ml), aqueous HCl and heated the reaction mass to 55-60° C. After completion of the reaction, the mass was dissolved in water, by adjusting pH with liquor ammonia. Filtered the solid and washed with water, dried the material at 45-50° C.
This free base was charged in 900 ml methanol and pH was adjusted to 1-1.5 by IPA.HCl and distilled off methanol completely to get white solid which was isolated by filtration.
Yield: 37 gm
HPLC Purity: 99.5%
Example-4: Preparation of [4-(2-Amino-1-Hydroxyethyl) Benzene-1, 2-Diol] (Racemic Norepinephrine Base)
Charged 2-amino-1-(3, 4-dihydroxyphenyl) ethanone hydrochloride (100 gm), 10% Pd/C(10 gm), methanol (700 ml) and water (300 ml) mixture in autoclave. Stirred the reaction mass at 40-45° C. After completion of reaction, Pd/C was removed by filtration. Collected filtrate and distilled off methanol. pH was adjusted by liquor ammonia. Isolated the solid by filtration and washed with water followed by methanol. Dried the solid at 40-45° C.
Yield: 67 gm
Purity: 99.2%
Example-5: Preparation of l-Norepinephrine Base
Charged racemic Norepinephrine base (100 gm), D-(−)-Tartaric acid (142 gm), water (100 ml) in a round bottom flask. The reaction mass was stirred to get clear solution. After some time, solid started to crystallize. Reaction mass was diluted with methanol (900 ml). Maintained the reaction mass under stirring for 24 hours at 25-30° C. Filtered and washed the wet cake with methanol to obtain Crude l-Norepinephrine tartrate salt.
Yield: 85 gm
The crude l-Norepinephrine tartrate salt was converted into its free base by dissolving this crude tartrate salt in water (500 ml) and adjusted pH to 8-8.5 by liquor ammonia and isolated the solid by filtration. Dried the material at 40-45° C. to obtain pure l-Norepinephrine free base (43 gm).
Yield: 43 gm (l-Norepinephrine pure base).
HPLC Purity: 99.7%
Chiral Purity: 98.0%
Example-6: Preparation of Pure l-Norepinephrine Base
Charged l-Norepinephrine base (100 gm) obtained from Example-5, D-(−)-Tartaric acid (142 gm), water (100 ml) in a round bottom flask. The reaction mass was stirred to get clear solution. After some time, a solid started to crystallize. Reaction mass was diluted with methanol (900 ml). Maintained the reaction mass under stirring for 24 hours at 25-30° C. Filtered and washed the wet cake with methanol to obtain l-Norepinephrine tartrate salt.
Yield: 88 gm
The l-Norepinephrine tartrate salt was converted into its free base by dissolving this crude tartrate salt in water (500 ml) and adjusted the pH to 8-8.5 by liquor ammonia and isolated the solid by filtration. Dried the material at 40-45° C. to obtain pure l-Norepinephrine free base (44 gm).
Yield: 44 gm (l-Norepinephrine pure base).
HPLC Purity: 99.7%
Chiral Purity: 99.1%
Example-7: Preparation of Highly Pure Norepinephrine Bitartrate Monohydrate
Charged Norepinephrine pure base (100 gm), L-(+) tartaric acid (100 gm), water (100 ml) and methanol (900 ml), Stirred the reaction mass to get clear solution. After some time, a solid started to crystallize then the reaction mass was diluted with methanol (900 ml). Maintained the reaction mass under stirring at 25-30° C. for 24 hours. Filtered and washed the wet cake with methanol to obtain Norepinephrine Bitartrate Monohydrate (90 gm).
HPLC Purity: 99.8%
Chiral Purity: 99.4%
Example-8: Purification of l-Norepinephrine Bitartrate Monohydrate
Charged 100 gm tartrate salt obtained from example-6, purified water (100 ml) and heated the reaction mass to 40-45° C. to obtain clear solution, cooled to 0-5° C. Charged IPA (100 ml) slowly and the mass was stirred for one hour. The solid was isolated by filtration and washed with IPA. Dried the material at 40-45° C. to obtain l-Norepinephrine Bitartrate Monohydrate (82 gm) having high enantiomeric purity.
HPLC Purity: 99.85%
Chiral Purity: 99.87%
Specific Optical rotation: −11.0°
Example-9
The following table sets forth the high purity of the l-Norepinephrine Bitartrate monohydrate of the invention as compared with prior art references.
[TABLE-US-00001] Referencel-Norepinephrine Example-2Bitartrate U.S. Pat. No.(JACS, 1948,monohydrate 2,774,789Page-2067-68,of the presentPurity CriteriaExample-AExample-a)invention Optical purity of l-68.45%77.14%99.87%NorepinephrineBitartratemonohydrateSpecific Optical−6.33°−10.4°−11.0°rotation(Limit: −10°to −12°)
It is evident from the above table that the compound of the present invention has substantially improved optical purity.
Publication numberPriority datePublication dateAssigneeTitleCN101053557A *2006-04-132007-10-17邵长青Noradrenaline bitartrate medicine composition frozen dried powder injectionCN102335123A *2010-07-162012-02-01上海禾丰制药有限公司Noradrenaline bitartrate injection and preparation technology thereofPublication numberPriority datePublication dateAssigneeTitleEP3110399B12014-02-272018-01-10Sintetica S.A.Process for producing a stable low concentration, injectable solution of noradrenalineFamily To Family CitationsCN109394683A *2018-12-072019-03-01远大医药(中国)有限公司A kind of preparation method of noradrenaline bitartrate injection
^ De Backer D, Biston P, Devriendt J, Madl C, Chochrad D, Aldecoa C, Brasseur A, Defrance P, Gottignies P, Vincent JL (March 2010). “Comparison of dopamine and norepinephrine in the treatment of shock”. The New England Journal of Medicine. 362 (9): 779–89. doi:10.1056/nejmoa0907118. PMID20200382.
^ I Moore, Joanne (6 December 2012). Pharmacology (3 ed.). Springer Science and Business Media. p. 39. ISBN9781468405248. Retrieved 19 November 2017.
A pyrimidine and thiazole derived ANTINEOPLASTIC AGENT and PROTEIN KINASE INHIBITOR of BCR-ABL KINASE. It is used in the treatment of patients with CHRONIC MYELOID LEUKEMIA who are resistant or intolerant to IMATINIB.
An orally bioavailable synthetic small molecule-inhibitor of SRC-family protein-tyrosine kinases. Dasatinib binds to and inhibits the growth-promoting activities of these kinases. Apparently because of its less stringent binding affinity for the BCR-ABL kinase, dasatinib has been shown to overcome the resistance to imatinib of chronic myeloid leukemia (CML) cells harboring BCR-ABL kinase domain point mutations. SRC-family protein-tyrosine kinases interact with a variety of cell-surface receptors and participate in intracellular signal transduction pathways; tumorigenic forms can occur through altered regulation or expression of the endogenous protein and by way of virally-encoded kinase genes. (NCI Thesaurus)
SPRYCEL (dasatinib) is an inhibitor of multiple tyrosine kinases.
The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2- methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate. The molecular formula is C22H26ClN7O2S • H2O, which corresponds to a formula weight of 506.02 (monohydrate).
The anhydrous free base has a molecular weight of 488.01. Dasatinib has the following chemical structure: Dasatinib is a white to off-white powder and has a melting point of 280°–286° C.
The drug substance is insoluble in water and slightly soluble in ethanol and methanol. SPRYCEL tablets are white to off-white, biconvex, film-coated tablets containing dasatinib, with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol
Drug Name:Dasatinib HydrateResearch Code:BMS-354825Trade Name:Sprycel®MOA:Kinase inhibitorIndication:Acute lymphoblastic leukaemia (ALL); Chronic myeloid leukemia (CML )Status:ApprovedCompany:Bristol-Myers Squibb (Originator)Sales:$1,620 Million (Y2015); $1,493 Million (Y2014); $1,280 Million (Y2013); $1,019 Million (Y2012); $803 Million (Y2011);ATC Code:L01XE06Approved Countries or Area
SPRYCEL (dasatinib) is a kinase inhibitor. The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate. The molecular formula is C22H26ClN7O2S • H2O, which corresponds to a formula weight of 506.02 (monohydrate). The anhydrous free base has a molecular weight of 488.01. Dasatinib has the following chemical structure:
Dasatinib is a white to off-white powder. The drug substance is insoluble in water and slightly soluble in ethanol and methanol.
SPRYCEL tablets are white to off-white, biconvex, film-coated tablets containing dasatinib, with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol.
Dasatinib hydrate was first approved by the U.S. Food and Drug Administration (FDA) on June 28, 2006, then approved by European Medicine Agency (EMA) on Nov 20, 2006, and approved by Pharmaceuticals and Medical Devices Agency of Japan (PMDA) on Jan 21, 2009. It was developed and marketed as Sprycel® by Bristol Myers Squibb in the US.
Dasatinibhydrate is a kinase inhibitor.It is indicated for the treatment ofchronic myeloid leukemia and acutelymphoblastic leukemia.
Sprycel® is available as film-coatedtabletfor oral use, containing 20, 50, 70, 80, 100 or 140 mg offreeDasatinib. The recommended dose is 100 mg once daily forchronic myeloid leukemia. Another dose is 140 mg once daily for accelerated phase chronic myeloid leukemia, myeloid or lymphoid blast phase chronic myeloid leukemia, or Ph+ acutelymphoblastic leukemia.
Dasatinib, also known as BMS-354825, is an orally bioavailable synthetic small molecule-inhibitor of SRC-family protein-tyrosine kinases. Dasatinib binds to and inhibits the growth-promoting activities of these kinases. Apparently because of its less stringent binding affinity for the BCR-ABL kinase, dasatinib has been shown to overcome the resistance to imatinib of chronic myeloid leukemia (CML) cells harboring BCR-ABL kinase domain point mutations.
In the EU dasatinib is indicated for children with
newly diagnosed Philadelphia chromosome-positive chronic myelogenous leukaemia in chronic phase (Ph+ CML CP) or Ph+ CML CP resistant or intolerant to prior therapy including imatinib.[2]
newly diagnosed Ph+ acute lymphoblastic leukaemia (ALL) in combination with chemotherapy.[2]
newly diagnosed Ph+ CML in chronic phase (Ph+ CML-CP) or Ph+ CML-CP resistant or intolerant to prior therapy including imatinib.[2]
and adults with
newly diagnosed Philadelphia-chromosome-positive (Ph+) chronic myelogenous leukaemia (CML) in the chronic phase;[2]
chronic, accelerated or blast phase CML with resistance or intolerance to prior therapy including imatinib mesilate;[2]
Ph+ acute lymphoblastic leukaemia (ALL) and lymphoid blast CML with resistance or intolerance to prior therapy.[2]
On October 11, 2011, the U.S. Food and Drug Administration (FDA) announced that dasatinib may increase the risk of a rare but serious condition in which there is abnormally high blood pressure in the arteries of the lungs (pulmonary hypertension, PAH).[9] Symptoms of PAH may include shortness of breath, fatigue, and swelling of the body (such as the ankles and legs).[9] In reported cases, people developed PAH after starting dasatinib, including after more than one year of treatment.[9] Information about the risk was added to the Warnings and Precautions section of the Sprycel drug label.[9]
Dasatinib is an ATP-competitive protein tyrosine kinase inhibitor. The main targets of dasatinib are BCR/Abl (the “Philadelphia chromosome”), Src, c-Kit, ephrin receptors, and several other tyrosine kinases.[11] Strong inhibition of the activated BCR-ABL kinase distinguishes dasatinib from other CML treatments, such as imatinib and nilotinib.[11][12] Although dasatinib only has a plasma half-life of three to five hours, the strong binding to BCR-ABL1 results in a longer duration of action.[12]
Dasatinib was developed by collaboration of Bristol-Myers Squibb and Otsuka Pharmaceutical Co., Ltd,[13][14][15] and named for Bristol-Myers Squibb research fellow Jagabandhu Das, whose program leader says that the drug would not have come into existence had he not challenged some of the medicinal chemists‘ underlying assumptions at a time when progress in the development of the molecule had stalled.[16]
Society and culture
Legal status
Dasatinib was approved for used in the United States in June 2006 and in the European Union in November 2006[17][2]
In October 2010, dasatinib was approved in the United States for the treatment of newly diagnosed adults with Philadelphia chromosome positive chronic myeloid leukemia in chronic phase (CP-CML).[18]
In November 2017, dasatinib was approved in the United States for the treatment of children with Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in the chronic phase.[19]
Approval was based on data from 97 pediatric participants with chronic phase CML evaluated in two trials—a Phase I, open-label, non-randomized, dose-ranging trial and a Phase II, open-label, non-randomized trial.[19] Fifty-one participants exclusively from the Phase II trial were newly diagnosed with chronic phase CML and 46 participants (17 from the Phase I trial and 29 from the Phase II trial) were resistant or intolerant to previous treatment with imatinib.[19] The majority of participants were treated with dasatinib tablets 60 mg/m2body surface area once daily.[19] Participants were treated until disease progression or unacceptable toxicity.[19]
Economics
The Union for Affordable Cancer Treatment objected to the price of dasatinib, in a letter to the U.S. trade representative. The average wholesale price in the U.S. is $367 per day, twice the price in other high income countries. The price in India, where the average annual per capita income is $1,570, and where most people pay out of pocket, is Rs6627 ($108) a day. Indian manufacturers offered to supply generic versions for $4 a day, but, under pressure from the U.S., the Indian Department of Industrial Policy and Promotion refused to issue a compulsory license.[20]
Bristol-Myers Squibb justified the high prices of cancer drugs with the high R&D costs, but the Union of Affordable Cancer Treatment said that most of the R&D costs came from the U.S. government, including National Institutes of Health funded research and clinical trials, and a 50% tax credit. In England and Wales, the National Institute for Health and Care Excellence recommended against dasatinib because of the high cost-benefit ratio.[20]
The Union for Affordable Cancer Treatment said that “the dasatinib dispute illustrates the shortcomings of US trade policy and its impact on cancer patients”[20]
Dasatinib has been shown to eliminate senescent cells in cultured adipocyte progenitor cells.[21] Dasatinib has been shown to induce apoptosis in senescent cells by inhibiting Src kinase, whereas quercetin inhibits the anti-apoptotic protein Bcl-xL.[21] Administration of dasatinib along with quercetin to mice improved cardiovascular function and eliminated senescent cells.[22] Aged mice given dasatinib with quercetin showed improved health and survival.[22]
Giving dasatinib and quercetin to mice eliminated senescent cells and caused a long-term resolution of frailty.[23] A study of fourteen human patients suffering from idiopathic pulmonary fibrosis (a disease characterized by increased numbers of senescent cells) given dasatinib and quercetin showed improved physical function and evidence of reduced senescent cells.[21]Route 1
Balaji, N.; Sultana, Sayeeda. Trace level determination and quantification of potential genotoxic impurities in dasatinib drug substance by UHPLC/infinity LC. International Journal of Pharmacy and Pharmaceutical Sciences. Department of Chemistry. St. Peter’s University. Tamil Nadu, India 600054. Volume 8. Issue 10. Pages 209-216. 2016
SYN 2
Reference
Zhang, Shaoning; Wei, Hongtao; Ji, Min. Synthesis of dasatinib. Zhongguo Yiyao Gongye Zazhi. Dept. of Pharmaceutical Engineering, School of Chemistry & Chemical Engineering. Southeast University. Nanjing, Jiangsu Province, Peop. Rep. China 210096. Volume 41. Issue 3. Pages 161-163. 2010
SYN 3
Reference
Suresh, Garbapu; Nadh, Ratnakaram Venkata; Srinivasu, Navuluri; Yennity, Durgaprasad. A convenient new and efficient commercial synthetic route for dasatinib (Sprycel). Synthetic Communications. Division of Chemistry, Department of Science and Humanities. Vignan’s Foundation for Science Technology and Research University. Guntur, India. Volume 47. Issue 17. Pages 1610-1621. 2017
SYN 4
Reference
Chen, Bang-Chi; Zhao, Rulin; Wang, Bei; Droghini, Roberto; Lajeunesse, Jean; Sirard, Pierre; Endo, Masaki; Balasubramanian, Balu; Barrish, Joel C. A new and efficient preparation of 2-aminothiazole-5-carbamides: applications to the synthesis of the anticancer drug dasatinib. ARKIVOC (Gainesville, FL, United States). Discovery Chemistry. Bristol-Myers Squibb Research and Development. Princeton, USA 08543. Issue 6.Pages 32-38. 2010
SYN 5
Reference
An, Kang; Guan, Jianning; Yang, Hao; Hou, Wen; Wan, Rong. Improvement on the synthesis of Dasatinib. Jingxi Huagong Zhongjianti. College of Science. Nanjing University of Technology. Nanjing, Jiangsu Province, Peop. Rep. China 211816. Volume 41. Issue 2. Pages 42-44. 2011
To a solution of S— sec-butyl-amine (7.31 g, 0.1 mol) in chloroform (80 mL) at 0° C. was slowly added benzoyl isothiocyanate (13.44 mL, 0.1 mol). The mixture was allowed to warm to 10° C. and stirred for 10 min. The solvent was then removed under reduced pressure, and the residue was dissolved in MeOH (80 mL). An aqueous solution (10 mL) of NaOH (4 g, 0.1 mol) was added to this solution, and the mixture was stirred at 60° C. for another 2 h. The MeOH was then removed under reduced pressure, and the residue was stirred in water (50 mL). The precipitate was collected by vacuum filtration and dried to provide S-1-sec-butyl-thiourea (12.2 g, 92% yield). mp 133-134° C.; 1H NMR (500 MHz, DMSO-D6) δ 7.40 (s, 1H), 7.20 (br s, 1H), 6.76 (s, 1H), 4.04 (s, 1H), 1.41 (m, 2H), 1.03 (d, J=6.1 Hz, 3H), 0.81 (d, J=7.7 Hz, 3H); 13C NMR (125 MHz, DMSO-D6) δ 182.5, 50.8, 28.8, 19.9, 10.3; LRMS m/z 133.2 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.49; H, 8.88; N, 21.32; S, 24.27.
Example 2Preparation of Intermediate:
(R)-1-sec-Butylthiourea
(R)-1-sec-Butylthiourea was prepared in 92% yield according to the general method outlined for Example 1. mp 133-134° C.; 1H NMR(500 MHz, DMSO) δ 0.80(m, 3H, J=7.7), 1.02(d, 3H, J=6.1), 1.41(m, 2H), (3.40, 4.04)(s, 1H), 6.76(s, 1H), 7.20(s, br, 1H), 7.39(d, 1H, J=7.2); 13C NMR (500 MHz, DMSO) δ: 10.00, 19.56, 28.50, 50.20, 182.00; m/z 133.23 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.32; H, 9.15; N, 21.14; S, 24.38.
Example 3Preparation of:
To a solution of 3-amino-N-methyl-4-methylbenzamide hydrochloride (1.0 g, 5 mmol) in acetone (10 mL) at 0° C. was added pyridine (1.2 mL, 15 mmol) dropwise via syringe. 3-Methoxyacryloyl chloride (0.72 mL. 6.5 mmol) was added and the reaction stirred at room temperature for 1 h. The solution was cooled again to 0° C. and 1N HCl (1.5 mL) was added dropwise via pipet. The reaction mixture was stirred for 5 min, then water (8.5 mL) was added via an addition funnel. The acetone was removed in vacuo and the resulting solution stirred for 4h. Crystallization began within 15 min. After stirring for 4 h, the vessel was cooled in an ice bath for 30 min, filtered, and rinsed with ice cold water (2×3 mL) to give compound 3A (0.99 g, 78% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.12 (br s, 1H), 7.76 (s, 1H), 7.29 (m, 2H), 7.05 (d, J=7.9 Hz, 1H), 5.47 (d, J=12.3 Hz, 1H), 3.48 (s, 3H), 2.54 (d, J=4.7 Hz, 3H), 2.03 (s, 3H); HPLC rt 2.28 min (Condition A).
3B. Example 3To a 50 mL RBF containing the above compound 3A (0.5 g, 2.0 mmol) was added THF (2.5 mL) and water (2 mL), followed by NBS (0.40 g, 2.22 mmol), and the solution was stirred for 90 min. R-sec-butylthiourea (Ex. 2) (267 mg), was added, and the solution was heated to 75° C. for 8 h. Conc. NH4OH was added to adjust the pH to 10 followed by the addition of EtOH (15 mL). Water (15 mL) was added and the slurry stirred for 16 h, filtered, and washed with water to give Example 3 as a light brown solid (0.48 g, 69% yield, 98% purity). MS 347.1; HPLC 2.59.
Example 4Preparation of:
Example 4 is prepared following the methods of Example 3 but using the appropriate acryl benzamide and Example 1.
Example 5Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (The Compound of Formula (IV))
5A. 1-(6-Chloro-2-methylpyrimidin-4-yl)thiourea
To a stirring slurry of 4-amino-5-chloro-2-methylpyrimidine (6.13 g, 42.7 mmol) in THF (24 mL) was added ethyl isothiocyanatoformate (7.5 mL, 63.6 mmol), and the mixture heated to reflux. After 5h, another portion of ethyl isothiocyanato formate (1.0 mL, 8.5 mmol) was added and after 10h, a final portion (1.5 mL, 12.7 mmol) was added and the mixture stirred 6h more. The slurry was evaporated under vacuum to remove most of the solvent and heptane (6 mL) added to the residue. The solid was collected by vacuum filtration and washed with heptane (2×5 mL) giving 8.01 g (68% yield) of the intermediate ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate.A solution of ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate (275 mg, 1.0 mmol) and 1N sodium hydroxide (3.5 eq) was heated and stirred at 50° C. for 2h. The resulting slurry was cooled to 20-22° C. The solid was collected by vacuum filtration, washed with water, and dried to give 185 mg of 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea (91% yield). 1H NMR (400 MHz, DMSO-d6): δ2.51 (S, 3H), 7.05 (s, 1H), 9.35 (s,1H), 10.07 (s, 1H), 10.91 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 25.25, 104.56, 159.19, 159.33, 167.36, 180.91.
To a cold stirring solution of 2-chloro-6-methylaniline (59.5 g 0.42 mol) and pyridine (68 ml, 0.63 mol) in THF (600 mL) was added 3-ethoxyacryloyl chloride (84.7 g, 0.63 mol) slowly keeping the temp at 0-5° C. The mixture was then warmed and stirred for 2 h. at 20° C. Hydrochloric acid (1N, 115 mL) was added at 0-10° C. The mixture was diluted with water (310 mL) and the resulting solution was concentrated under vacuum to a thick slurry. The slurry was diluted with toluene (275 mL) and stirred for 15 min. at 20-22° C. then 1 h. at 0° C. The solid was collected by vacuum filtration, washed with water (2×75 mL) and dried to give 74.1 g (73.6% yield) of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide). 1H NMR (400 Hz, DMSO-d6) δ 1.26 (t, 3H, J=7 Hz), 2.15 (s, 3H), 3.94 (q, 2H, J=7 Hz), 5.58 (d, 1H, J=12.4 Hz), 7.10-7.27 (m, 2H, J=7.5 Hz), 7.27-7.37 (d, 1H, J=7.5 Hz), 7.45(d, 1H, J=12.4 Hz), 9.28 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 14.57, 18.96, 67.17, 97.99, 126.80, 127.44, 129.07, 131.32, 132.89, 138.25, 161.09, 165.36.
To a mixture of compound 5B (5.00 g, 20.86 mmol) in 1,4-dioxane (27 mL) and water (27 mL) was added NBS (4.08 g, 22.9 mmol) at −10 to 0° C. The slurry was warmed and stirred at 20-22° C. for 3h. Thiourea (1.60 g, 21 mmol) was added and the mixture heated to 80° C. After 2h, the resulting solution was cooled to 20-22° and conc. ammonium hydroxide (4.2 mL) was added dropwise. The resulting slurry was concentrated under vacuum to about half volume and cooled to 0-5° C. The solid was collected by vacuum filtration, washed with cold water (10 mL), and dried to give 5.3 g (94.9% yield) of 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6) δ δ 2.19 (s, 3H), 7.09-7.29 (m, 2H, J=7.5), 7.29-7.43 (d, 1H, J=7.5), 7.61 (s, 2H), 7.85 (s, 1H), 9.63 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 18.18, 120.63, 126.84, 127.90, 128.86, 132.41, 133.63, 138.76, 142.88, 159.45, 172.02.
To a stirring solution of compound 5C (5.00 g, 18.67 mmol) and 4,6-dichloro-2-methylpyrimidine (3.65 g 22.4/mmol) in THF (65 mL) was added a 30% wt. solution of sodium t-butoxide in THF (21.1 g, 65.36 mmol) slowly with cooling to keep the temperature at 10-20° C. The mixture was stirred at room temperature for 1.5 h and cooled to 0-5° C. Hydrochloric acid, 2N (21.5 mL) was added slowly and the mixture stirred 1.75 h at 0-5° C. The solid was collected by vacuum filtration, washed with water (15 mL) and dried to give 6.63 g (86.4% yield) of compound 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).
5E. Example 5To a mixture of compound 5D (4.00 g, 10.14 mmol) and hydroxyethylpiperazine (6.60 g, 50.69 mmol) in n-butanol (40 mL) was added DIPEA (3.53 mL, 20.26 mmol). The slurry was heated at 118° C. for 4.5 h, then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with n-butanol (5 mL), and dried. The product (5.11 g) was dissolved in hot 80% EtOH—H2O (80 mL), and the solution was clarified by filtration. The hot solution was slowly diluted with water (15 mL) and cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with 50% ethanol-water (5 mL) and dried affording 4.27 g (83.2% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide as monohydrate. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.40 (s, 3H), 2.42 (t, 2H, J=6), 2.48 (t, 4H, J=6.3), 3.50 (m, 4H), 3.53 (q, 2H, J=6), 4.45 (t, 1H, J=5.3), 6.04 (s, 1H), 7.25 (t, 1H, J=7.6), 7.27 (dd, 1H, J=7.6, 1.7), 7.40 (dd, 1H, J=7.6, 1.7), 8.21 (s, 1H), 9.87 (s, 1H), 11.47.
To a slurry of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide 5B (120 mg, 0.50 mmol) in THF (0.75 ml) and water (0.5 mL) was added NBS (98 mg, 0.55 mmol) at 0° C. The mixture was warmed and stirred at 20-22° C. for 3h. To this was added 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea 5A (100 mg, 0.49 mmol), and the slurry heated and stirred at reflux for 2h. The slurry was cooled to 20-22° C. and the solid collected by vacuum filtration giving 140 mg (71% yield) of 2-(6-chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).Compound 5D was elaborated to N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide, following Step 5E.
2-piperazin-1-yl-ethanol (8.2 g, 63.1 mmol) was added to a solution of 4,6-dichloro-2-methylpyrimidine (5.2 g, 31.9 mmol) in dichloromethane (80 ml) at rt. The mixture was stirred for two hours and triethylamine (0.9 ml) was added. The mixture was stirred at rt for 20h. The resultant solid was filtered. The cake was washed with dichloromethane (20 ml). The filtrate was concentrated to give an oil. This oil was dried under high vacuum for 20h to give a solid. This solid was stirred with heptane (50 ml) at rt for 5h. Filtration gave 7C (8.13 g) as a white solid
7B. Example 7
To a 250 ml of round bottom flask were charged compound 5C (1.9 g, 7.1 mmol), compound 7C (1.5 g, 5.9 mmol), K2CO3 (16 g, 115.7 mmol), Pd (OAc)2 (52 mg, 0.23 mmol) and BINAP (291 mg, 0.46 mmol). The flask was placed under vacuum and flushed with nitrogen. Toluene was added (60 ml). The suspension was heated to 100-110° C. and stirred at this temperature for 20h. After cooling to room temperature, the mixture was applied to a silica gel column. The column was first eluted with EtOAC, and then with 10% of MeOH in EtOAC. Finally, the column was washed with 10% 2M ammonia solution in MeOH/90% EtOAC. The fractions which contained the desired product were collected and concentrated to give compound IV as a yellow solid (2.3 g).
Analytical MethodsSolid State Nuclear Magnetic Resonance (SSNMR)All solid-state C-13 NMR measurements were made with a Bruker DSX-400, 400 MHz NMR spectrometer. High resolution spectra were obtained using high-power proton decoupling and the TPPM pulse sequence and ramp amplitude cross-polarization (RAMP-CP) with magic-angle spinning (MAS) at approximately 12 kHz (A. E. Bennett et al, J. Chem. Phys., 1995, 103, 6951), (G. Metz, X. Wu and S. O. Smith, J. Magn. Reson. A., 1994, 110, 219-227). Approximately 70 mg of sample, packed into a canister-design zirconia rotor was used for each experiment. Chemical shifts (δ) were referenced to external adamantane with the high frequency resonance being set to 38.56 ppm (W. L. Earl and D. L. VanderHart, J. Magn. Reson., 1982, 48, 35-54).X-Ray Powder DiffractionOne of ordinary skill in the art will appreciate that an X-ray diffraction pattern may be obtained with a measurement error that is dependent upon the measurement conditions employed. In particular, it is generally known that intensities in a X-ray diffraction pattern may fluctuate depending upon measurement conditions employed. It should be further understood that relative intensities may also vary depending upon experimental conditions and, accordingly, the exact order of intensity should not be taken into account. Additionally, a measurement error of diffraction angle for a conventional X-ray diffraction pattern is typically about 5% or less, and such degree of measurement error should be taken into account as pertaining to the aforementioned diffraction angles. Consequently, it is to be understood that the crystal forms of the instant invention are not limited to the crystal forms that provide X-ray diffraction patterns completely identical to the X-ray diffraction patterns depicted in the accompanying Figures disclosed herein. Any crystal forms that provide X-ray diffraction patterns substantially identical to those disclosed in the accompanying Figures fall within the scope of the present invention. The ability to ascertain substantial identities of X-ray diffraction patterns is within the purview of one of ordinary skill in the art.X-Ray powder diffraction data for the crystalline forms of Compound (IV) were obtained using a Bruker GADDS (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) (General Area Detector Diffraction System) manual chi platform goniometer. Powder samples were placed in thin walled glass capillaries of 1 mm or less in diameter; the capillary was rotated during data collection. The sample-detector distance was 17 cm. The radiation was Cu Kα (45 kV 111 mA, λ=1.5418 Å). Data were collected for 3<2θ<35° with a sample exposure time of at least 300 seconds.Single Crystal X-RayAll single crystal data were collected on a Bruker-Nonius (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) Kappa CCD 2000 system using Cu Kα radiation (λ=1.5418 Å) and were corrected only for the Lorentz-polarization factors. Indexing and processing of the measured intensity data were carried out with the HKL2000 software package (Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W. C. Jr & Sweet, R. M. (Academic, NY), Vol. 276, pp. 307-326) in the Collect program suite (Data collection and processing user interface: Collect: Data collection software, R. Hooft, Nonius B. V., 1998).The structures were solved by direct methods and refined on the basis of observed reflections using either the SDP (SDP, Structure Determination Package, Enraf-Nonius, Bohemia NY 11716 Scattering factors, including f′ and f″, in the SDP software were taken from the “International Tables for Crystallography”, Kynoch Press, Birmingham, England, 1974; Vol IV, Tables 2.2A and 2.3.1) software package with minor local modifications or the crystallographic package, MAXUS (maXus solution and refinement software suite: S. Mackay, C. J. Gilmore, C. Edwards, M. Tremayne, N. Stewart, K. Shankland. maXus: a computer program for the solution and refinement of crystal structures from diffraction data).The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were varied.The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were variedDifferential Scanning CalorimetryThe DSC instrument used to test the crystalline forms was a TA Instruments® model Q1000. The DSC cell/sample chamber was purged with 100 ml/min of ultra-high purity nitrogen gas. The instrument was calibrated with high purity indium. The accuracy of the measured sample temperature with this method is within about +/−1° C., and the heat of fusion can be measured within a relative error of about +/−5%. The sample was placed into an open aluminum DSC pan and measured against an empty reference pan. At least 2 mg of sample powder was placed into the bottom of the pan and lightly tapped down to ensure good contact with the pan. The weight of the sample was measured accurately and recorded to a hundredth of a milligram. The instrument was programmed to heat at 10° C. per minute in the temperature range between 25 and 350° C.The heat flow, which was normalized by a sample weight, was plotted versus the measured sample temperature. The data were reported in units of watts/gram (“W/g”). The plot was made with the endothermic peaks pointing down. The endothermic melt peak was evaluated for extrapolated onset temperature, peak temperature, and heat of fusion in this analysis.Thermogravimetric Analysis (TGA)The TGA instrument used to test the crystalline forms was a TAInstruments® model Q500. Samples of at least 10 milligrams were analyzed at a heating rate of 10° C. per minute in the temperature range between 25° C. and about 350° C.
Example 8Preparation of:
crystalline monohydrate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)An example of the crystallization procedure to obtain the crystalline monohydrate form is shown here:
Charge 48 g of the compound of formula (IV).
Charge approximately 1056 mL (22 mL/g) of ethyl alcohol, or other suitable alcohol.
Charge approximately 144 mL of water.
Dissolve the suspension by heating to approximately 75° C.
Optional: Polish filter by transfer the compound of formula (IV) solution at 75° C. through the preheated filter and into the receiver.
Rinse the dissolution reactor and transfer lines with a mixture of 43 mL of ethanol and 5 mL of water.
Heat the contents in the receiver to 75-80° C. and maintain 75-80° C. to achieve complete dissolution.Charge approximately 384 mL of water at a rate such that the batch temperature is maintained between 75-80° C.Cool to 75° C., and, optionally, charge monohydrate seed crystals. Seed crystals are not essential to obtaining monohydrate, but provide better control of the crystallization.
Cool to 70° C. and maintain 70° C. for ca. 1 h.
Cool from 70 to 5 C over 2 h, and maintain the temperature between 0 at 5° C. for at least 2 h.
Filter the crystal slurry.
Wash the filter cake with a mixture of 96 mL of ethanol and 96 mL of water.
Dry the material at ≦50° C. under reduced pressure until the water content is 3.4 to 4.1% by KF to afford 41 g (85 M %). Alternately, the monohydrate can be obtained by:
1) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate and heated at 80° C. to give bulk monohydrate.
2) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate. On standing several days at room temperature, bulk monohydrate had formed.
3) An aqueous suspension of compound IV was seeded with monohydrate and heated at 70° C. for 4 hours to give bulk monohydrate. In the absence of seeding, an aqueous slurry of compound IV was unchanged after 82 days at room temperature.
4) A solution of compound IV in a solvent such as NMP or DMA was treated with water until the solution became cloudy and was held at 75-85° C. for several hours. Monohydrate was isolated after cooling and filtering.
5) A solution of compound IV in ethanol, butanol, and water was heated. Seeds of monohydrate were added to the hot solution and then cooled. Monohydrate was isolated upon cooling and filtration.
One of ordinary skill in the art will appreciate that the monohydrate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 1 or by a representative sampling of peaks as shown in Table 1.Representative peaks taken from the XRPD of the monohydrate of the compound of formula (IV) are shown in Table 1.TABLE 1 2-Theta d(Å) Height 17.994 4.9257 915 18.440 4.8075 338 19.153 4.6301 644 19.599 4.5258 361 21.252 4.1774 148 24.462 3.6359 250 25.901 3.4371 133 28.052 3.1782 153The XRPD is also characterized by the following list comprising 2θ values selected from the group consisting of: 4.6±0.2, 11.2±0.2, 13.8±0.2, 15.2±0.2, 17.9±0.2, 19.1±0.2, 19.6±0.2, 23.2±0.2, 23.6±0.2. The XRPD is also characterized by the list of 2θ values selected from the group consisting of: 18.0±0.2, 18.4±0.2, 19.2±0.2, 19.6±0.2, 21.2±0.2, 24.5±0.2, 25.9±0.2, and 28.0±0.2.Single crystal x-ray data was obtained at room temperature (+25° C.). The molecular structure was confirmed as a monohydrate form of the compound of Formula (IV).The following unit cell parameters were obtained for the monohydrate of the compound of formula (IV) from the x-ray analysis at 25° C.:a(Å)=13.8632(7); b(Å)=9.3307(3); c(Å)=38.390(2);V(Å3) 4965.9(4); Z′=1; Vm=621Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.354Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).Single crystal x-ray data was also obtained at −50° C. The monohydrate form of the compound of Formula (IV) is characterized by unit cell parameters approximately equal to the following:Cell dimensions:
a(Å)=13.862(1);
b(Å)=9.286(1);
c(Å)=38.143(2);
Volume=4910(1) Å3Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.369wherein the compound is at a temperature of about −50° C.The simulated XRPD was calculated from the refined atomic parameters at room temperature.The monohydrate of the compound of formula (IV) is represented by the DSC as shown in FIG. 2. The DSC is characterized by a broad peak between approximately 95° C. and 130° C. This peak is broad and variable and corresponds to the loss of one water of hydration as seen in the TGA graph. The DSC also has a characteristic peak at approximately 287° C. which corresponds to the melt of the dehydrated form of the compound of formula (IV).The TGA for the monohydrate of the compound of Formula (IV) is shown in FIG. 2 along with the DSC. The TGA shows a 3.48% weight loss from 50° C. to 175° C. The weight loss corresponds to a loss of one water of hydration from the compound of Formula (IV).The monohydrate may also be prepared by crystallizing from alcoholic solvents, such as methanol, ethanol, propanol, i-propanol, butanol, pentanol, and water.
Example 9Preparation of:
crystalline n-butanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)The crystalline butanol solvate of the compound of formula (IV) is prepared by dissolving compound (IV) in 1-butanol at reflux (116-118° C.) at a concentration of approximately 1 g/25 mL of solvent. Upon cooling, the butanol solvate crystallizes out of solution. Filter, wash with butanol, and dry.The following unit cell parameters were obtained from the x-ray analysis for the crystalline butanol solvate, obtained at room temperature:a(Å)=22.8102(6); b(Å)=8.4691(3); c(Å)=15.1436(5); β=95.794(2);V(Å3) 2910.5(2); Z′=1; Vm=728Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.283Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the butanol solvate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 3 or by a representative sampling of peaks. Representative peaks for the crystalline butanol solvate are 2θ values of: 5.9±0.2, 12.0±0.2, 13.0±0.2, 17.7±0.2, 24.1±0.2, and 24.6±0.2.
Example 10Preparation of:
crystalline ethanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)
To a 100-mL round bottom flask was charged 4.00 g (10.1 mmol) of 5D (contained 2.3 Area % 5C) 6.60 g (50.7 mmol) of 7B, 80 mL of n-butanol and 2.61 g (20.2 mmol) of DIPEA. The resulting slurry was heated to 120° C. and maintained at 120° C. for 4.5 h whereby HPLC analysis showed 0.19 relative Area % of residual 5D to compound IV. The homogeneous mixture was cooled to 20° C. and left stirring overnight. The resulting crystals were filtered. The wet cake was washed twice with 10-mL portions of n-butanol to afford a white crystalline product. HPLC analysis showed this material to contain 99.7 Area % compound IV and 0.3 Area % 5C.The resulting wet cake was returned to the 100-mL reactor, and charged with 56 mL (12 mL/g) of 200 proof ethanol. At 80° C. an additional 25 mL of ethanol was added. To this mixture was added 10 mL of water resulting in rapid dissolution. Heat was removed and crystallization was observed at 75-77° C. The crystal slurry was further cooled to 20° C. and filtered. The wet cake was washed once with 10 mL of 1:1 ethanol: water and once with 10 mL of n-heptane. The wet cake contained 1.0% water by KF and 8.10% volatiles by LOD. The material was dried at 60° C./30 in Hg for 17 h to afford 3.55 g (70 M %) of material containing only 0.19% water by KF, 99.87 Area % by HPLC. The 1H NMR spectrum, however revealed that the ethanol solvate had been formed.The following unit cell parameters were obtained from the x-ray analysis for the crystalline ethanol solvate (di-ethanolate, E2-1), obtained at −40° C.:a(Å)=22.076(1); b(Å)=8.9612(2); c(Å)=16.8764(3); β=114.783(1);V(Å3) 3031.1(1); Z′=1; Vm=758Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.271Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 4 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 5.8±0.2, 11.3±0.2, 15.8±0.2, 17.2±0.2, 19.5±0.2, 24.1±0.2, 25.3±0.2, and 26.2±0.2.In addition, during the process to form the ethanolate (diethanolate) the formation of another ethanol solvate (½ ethanolate, T1E2-1) has been observed. To date this additional ethaonol solvate is known strictly as a partial desolvation product of the original diethanolate form E2-1, and has only been observed on occasion during crystallization of E2-1The following unit cell parameters were obtained from the x-ray analysis for the crystalline ½ ethanol solvate T1E2-1, obtained at −10° C.:a(Å)=22.03(2); b(Å)=9.20(1); c(Å)=12.31(1);β=93.49(6)V(Å3) 2491(4)); Z′=1; Vm=623;Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.363Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (T1E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 7 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 7.20±0.2, 12.01±0.2, 12.81±0.2, 18.06±0.2, 19.30±0.2, and 25.24±0.2.
Example 11Preparation of:
crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (Neat form N-6)To a mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL) was added DIPEA (155 mL, 0.89 mol). The suspension was heated at 110° C. (solution obtained) for 25 min., then cooled to about 90° C. The resulting hot solution was added dropwise into hot (80° C.) water (8010) mL, keeping the temperature at about 80° C. The resulting suspension was stirred 15 min at 80° C. then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with water (2×1600 mL) and dried in vacuo at 55-60° C. affording 192.45 g (88.7% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H), 2.41 (s, 3H), 2.43 (t, 2H, J=6), 2.49 (t, 4H, J=6.3), 3.51 (m, 4H), 3.54 (q, 2H, J=6), 4.46 (t, 1H, J=5.3), 6.05 (s, 1H), 7.26 (t, 1H, J=7.6), 7.28 (dd, 1H, J=7.6, 1.7), 7.41 (dd, 1H, J=7.6, 1.7), 8.23 (s, 1H), 9.89 (s, 1H), 11.48. KF0.84; DSC: 285.25° C. (onset), 286.28° C. (max).The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline compound IV, obtained at 23° C.:a(Å)=22.957(1); b(Å)=8.5830(5); c(Å)=13.803(3); β=112.039(6);V(Å3)=2521.0(5); Z′=1; Vm=630Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.286Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the crystalline form of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 5 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (N-6) are 2θ values of: 6.8±0.2, 11.1±0.2, 12.3±0.2, 13.2±0.2, 13.7±0.2, 16.7±0.2, 21.0±0.2, 24.3±0.2, and 24.8±0.2.
Example 12Preparation of:
crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (neatform T1H1-7)The title neat form may be prepared by heating the monohydrate form of the compound of formula (IV) above the dehydration temperature.The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline (T1H1-7) compound IV, obtained at 25° C.:a(Å)=13.4916; b(Å)=9.3992(2); c(Å)=38.817(1);V(Å3)=4922.4(3); Z′=1; Vm=615Space group PbcaDensity (calculated) (g/cm3) 1.317Wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the neat crystalline form (T1H1-7) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 6 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (T1H1-7)) are 2θ values of: 8.0±0.2, 9.7±0.2, 11.2±0.2, 13.3±0.2, 17.5±0.2, 18.9±0.2, 21.0±0.2, 22.0±0.2.Obviously, numerous modifications and variations of the present invention are possible in light of the above teachings. It is therefore to be understood that within the scope of the appended claims, the invention may be practiced otherwise than as specifically described herein.PATENThttps://patents.google.com/patent/US8680103B2/enAminothiazole-aromatic amides of formula I
wherein Ar is aryl or heteroaryl, L is an optional alkylene linker, and R2, R3, R4, and R5, are as defined in the specification herein, are useful as kinase inhibitors, in particular, inhibitors of protein tyrosine kinase and p38 kinase. They are expected to be useful in the treatment of protein tyrosine kinase-associated disorders such as immunologic and oncological disorders [see, U.S. Pat. No. 6,596,746 (the ‘746 patent), assigned to the present assignee and incorporated herein by reference], and p38 kinase-associated conditions such as inflammatory and immune conditions, as described in U.S. patent application Ser. No. 10/773,790, filed Feb. 6, 2004, claiming priority to U.S. Provisional application Ser. No. 60/445,410, filed Feb. 6, 2003 (hereinafter the ‘410 application), both of which are also assigned to the present assignee and incorporated herein by reference.The compound of formula (IV), ′N-(2-Chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, is an inhibitor of SRC/ABL and is useful in the treatment of oncological diseases.
Other approaches to preparing 2-aminothiazole-5-carboxamides are described in the ‘746 patent and in the ‘410 application. The ‘746 patent describes a process involving treatment of chlorothiazole with n-BuLi followed by reaction with phenyl isocyanates to give chlorothiazole-benzamides, which are further elaborated to aminothiazole-benzamide final products after protection, chloro-to-amino substitution, and deprotection, e.g.,
The ‘410 application describes a multi-step process involving first, converting N-unsubstituted aminothiazole carboxylic acid methyl or ethyl esters to bromothiazole carboxylic acid esters via diazotization with tert-butyl nitrite and subsequent CuBr2 treatment, e.g.,
then, hydrolyzing the resulting bromothiazole esters to the corresponding carboxylic acids and converting the acids to the corresponding acyl chlorides, e.g.,
then finally, coupling the acyl chlorides with anilines to afford bromothiazole-benzamide intermediates which were further elaborated to aminothiazole-benzamide final products, e.g.,
Other approaches for making 2-aminothiazole-5-carboxamides include coupling of 2-aminothiazole-5-carboxylic acids with amines using various coupling conditions such as DCC [Roberts et al, J. Med. Chem. (1972), 15, at p. 1310], and DPPA [Marsham et al., J. Med. Chem. (1991), 34, at p. 1594)].The above methods present drawbacks with respect to the production of side products, the use of expensive coupling reagents, less than desirable yields, and the need for multiple reaction steps to achieve the 2-aminothiazole-5-carboxamide compounds.Reaction of N,N-dimethyl-N′-(aminothiocarbonyl)-formamidines with α-haloketones and esters to give 5-carbonyl-2-aminothiazoles has been reported. See Lin, Y. et al, J. Heterocycl. Chem. (1979), 16, at 1377; Hartmann, H. et al, J. Chem. Soc. Perkin Trans. (2000), 1, at 4316; Noack, A. et al; Tetrahedron (2002), 58, at 2137; Noack, A.; et al. Angew. Chem. (2001), 113, at 3097; and Kantlehner, W. et al., J. Prakt. Chem./Chem.-Ztg. (1996), 338, at 403. Reaction of β-ethoxy acrylates and thioureas to prepare 2-aminothiazole-5-carboxylates also has been reported. See Zhao, R., et al., Tetrahedron Lett. (2001), 42, at 2101. However, electrophilic bromination of acrylanilide and crotonanilide has been known to undergo both aromatic bromination and addition to the α,β-unsaturated carbon-carbon double bonds. See Autenrieth, Chem. Ber. (1905), 38, at 2550; Eremeev et al., Chem. Heterocycl. Compd. Engl. Transl. (1984), 20, at 1102.New and efficient processes for preparing 2-aminothiazole-5-carboxamides are desired.
SUMMARY OF THE INVENTION
This invention is related to processes for the preparation of 2-aminothiazole-5-aromatic amides having the formula (I),
wherein L, Ar, R2, R3, R4, R5, and m are as defined below, comprising reacting a compound having the formula (II),
wherein Q is the group —O—P*, wherein P* is selected so that, when considered together with the oxygen atom to which P* is attached, Q is a leaving group, and Ar, L, R2, R3, and m are as defined below, with a halogenating reagent in the presence of water followed by a thiourea compound having the formula (III),
wherein, R4 and R5 are as defined below, to provide the compound of formula (I),
wherein,Ar is the same in formulae (I) and (II) and is aryl or heteroaryl;L is the same in formulae (I) and (II) and is optionally-substituted alkylene;R2 is the same in formulae (I) and (II), and is selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo;R3 is the same in formulae (I) and (II), and is selected from hydrogen, halogen, cyano, haloalkyl, alkyl, substituted alkyl, alkenyl, substituted alkenyl, aryl, heteroaryl, cycloalkyl, and heterocyclo;R4 is (i) the same in each of formulae (I) and (III), and (ii) is independently selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo, or alternatively, R4 is taken together with R5, to form heteroaryl or heterocyclo;R5 is (i) the same in each of formulae (I) and (III), and (ii) is independently selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, alkynyl, substituted alkynyl, aryl, heteroaryl, cycloalkyl, and heterocyclo, or alternatively, R5 is taken together with R4, to form heteroaryl or heterocyclo; andm is 0 or 1.Applicants have surprisingly discovered said process for converting β-(P*)oxy acryl aromatic amides and thioureas to 2-aminothiazole derivatives, wherein the aromatic amides are not subject to further halogenation producing other side products. Aminothiazole-aromatic amides, particularly, 2-aminothiazole-5-benzamides, can thus be efficiently prepared with this process in high yield.In another aspect, the present invention is directed to crystalline forms of the compound of formula (IV).
EXAMPLESExample 1Preparation of Intermediate:
(S)-1-sec-Butylthiourea
To a solution of S-sec-butyl-amine (7.31 g, 0.1 mol) in chloroform (80 mL) at 0° C. was slowly added benzoyl isothiocyanate (13.44 mL, 0.1 mol). The mixture was allowed to warm to 10° C. and stirred for 10 min. The solvent was then removed under reduced pressure, and the residue was dissolved in MeOH (80 mL). An aqueous solution (10 mL) of NaOH (4 g, 0.1 mol) was added to this solution, and the mixture was stirred at 60° C. for another 2 h. The MeOH was then removed under reduced pressure, and the residue was stirred in water (50 mL). The precipitate was collected by vacuum filtration and dried to provide S-1-sec-butyl-thiourea (12.2 g, 92% yield). mp 133-134° C.; 1H NMR (500 MHz, DMSO-D6) δ 7.40 (s, 1H), 7.20 (br s, 1H), 6.76 (s, 1H), 4.04 (s, 1H), 1.41 (m, 2H), 1.03 (d, J=6.1 Hz, 3H), 0.81 (d, J=7.7 Hz, 3H); 13C NMR (125 MHz, DMSO-D6) δ 182.5, 50.8, 28.8, 19.9, 10.3; LRMS m/z 133.2 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.49; H, 8.88; N, 21.32; S, 24.27.
Example 2Preparation of Intermediate:
(R)-1-sec-Butylthiourea
(R)-1-sec-Butylthiourea was prepared in 92% yield according to the general method outlined for Example 1. mp 133-134° C.; 1H NMR (500 MHz, DMSO) δ 0.80 (m, 3H, J=7.7), 1.02 (d, 3H, J=6.1), 1.41 (m, 2H), (3.40, 4.04) (s, 1H), 6.76 (s, 1H), 7.20 (s, br, 1H), 7.39 (d, 1H, J=7.2); 13C NMR (500 MHz, DMSO) δ: 10.00, 19.56, 28.50, 50.20, 182.00; m/z 133.23 (M+H); Anal. Calcd for C5H12N2S: C, 45.41; H, 9.14; N, 21.18; S, 24.25. Found: C, 45.32; H, 9.15; N, 21.14; S, 24.38.
Example 3Preparation of:
To a solution of 3-amino-N-methyl-4-methylbenzamide hydrochloride (1.0 g, 5 mmol) in acetone (10 mL) at 0° C. was added pyridine (1.2 mL, 15 mmol) dropwise via syringe. 3-Methoxyacryloyl chloride (0.72 mL 6.5 mmol) was added and the reaction stirred at room temperature for 1 h. The solution was cooled again to 0° C. and 1N HCl (1.5 mL) was added dropwise via pipette. The reaction mixture was stirred for 5 min, then water (8.5 mL) was added via an addition funnel. The acetone was removed in vacuo and the resulting solution stirred for 4 h. Crystallization began within 15 min. After stirring for 4 h, the vessel was cooled in an ice bath for 30 min, filtered, and rinsed with ice cold water (2×3 mL) to give compound 3A (0.99 g, 78% yield) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.95 (s, 1H), 8.12 (br s, 1H), 7.76 (s, 1H), 7.29 (m, 2H), 7.05 (d, J=7.9 Hz, 1H), 5.47 (d, J=12.3 Hz, 1H), 3.48 (s, 3H), 2.54 (d, J=4.7 Hz, 3H), 2.03 (s, 3H); HPLC rt 2.28 min (Condition A).
3B. Example 3To a 50 mL RBF containing the above compound 3A (0.5 g, 2.0 mmol) was added THF (2.5 mL) and water (2 mL), followed by NBS (0.40 g, 2.22 mmol), and the solution was stirred for 90 min. R-sec-butylthiourea (Ex. 2) (267 mg), was added, and the solution was heated to 75° C. for 8 h. Conc. NH4OH was added to adjust the pH to 10 followed by the addition of EtOH (15 mL). Water (15 mL) was added and the slurry stirred for 16 h, filtered, and washed with water to give Example 3 as a light brown solid (0.48 g, 69% yield, 98% purity). MS 347.1; HPLC 2.59.
Example 4Preparation of:
Example 4 is prepared following the methods of Example 3 but using the appropriate acryl benzamide and Example 1.
Example 5Preparation of:
N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (The compound of Formula (IV))
5A. 1-(6-Chloro-2-methylpyrimidin-4-yl)thiourea
To a stirring slurry of 4-amino-5-chloro-2-methylpyrimidine (6.13 g, 42.7 mmol) in THF (24 mL) was added ethyl isothiocyanatoformate (7.5 mL, 63.6 mmol), and the mixture heated to reflux. After 5 h, another portion of ethyl isothiocyanato formate (1.0 mL, 8.5 mmol) was added and after 10 h, a final portion (1.5 mL, 12.7 mmol) was added and the mixture stirred 6 h more. The slurry was evaporated under vacuum to remove most of the solvent and heptane (6 mL) added to the residue. The solid was collected by vacuum filtration and washed with heptane (2×5 mL) giving 8.01 g (68% yield) of the intermediate ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate.A solution of ethyl 6-chloro-2-methylpyrimidin-4-ylcarbamothioylcarbamate (275 mg, 1.0 mmol) and 1N sodium hydroxide (3.5 eq) was heated and stirred at 50° C. for 2 h. The resulting slurry was cooled to 20-22° C. The solid was collected by vacuum filtration, washed with water, and dried to give 185 mg of 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea (91% yield). 1H NMR (400 MHz, DMSO-d6): δ2.51 (S, 3H), 7.05 (s, 1H), 9.35 (s, 1H), 10.07 (s, 1H), 10.91 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 25.25, 104.56, 159.19, 159.33, 167.36, 180.91.
To a cold stirring solution of 2-chloro-6-methylaniline (59.5 g 0.42 mol) and pyridine (68 ml, 0.63 mol) in THF (600 mL) was added 3-ethoxyacryloyl chloride (84.7 g, 0.63 mol) slowly keeping the temp at 0-5° C. The mixture was then warmed and stirred for 2 h. at 20° C. Hydrochloric acid (1N, 115 mL) was added at 0-10° C. The mixture was diluted with water (310 mL) and the resulting solution was concentrated under vacuum to a thick slurry. The slurry was diluted with toluene (275 mL) and stirred for 15 min. at 20-22° C. then 1 h. at 0° C. The solid was collected by vacuum filtration, washed with water (2×75 mL) and dried to give 74.1 g (73.6% yield) of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide). 1H NMR (400 Hz, DMSO-d6) δ 1.26 (t, 3H, J=7 Hz), 2.15 (s, 3H), 3.94 (q, 2H, J=7 Hz), 5.58 (d, 1H, J=12.4 Hz), 7.10-7.27 (m, 2H, J=7.5 Hz), 7.27-7.37 (d, 1H, J=7.5 Hz), 7.45 (d, 1H, J=12.4 Hz), 9.28 (s, 1H); 13C NMR (100 MHz, CDCl3) δ: 14.57, 18.96, 67.17, 97.99, 126.80, 127.44, 129.07, 131.32, 132.89, 138.25, 161.09, 165.36.
To a mixture of compound 5B (5.00 g, 20.86 mmol) in 1,4-dioxane (27 mL) and water (27 mL) was added NBS (4.08 g, 22.9 mmol) at −10 to 0° C. The slurry was warmed and stirred at 20-22° C. for 3 h. Thiourea (1.60 g, 21 mmol) was added and the mixture heated to 80° C. After 2 h, the resulting solution was cooled to 20-22° and conc. ammonium hydroxide (4.2 mL) was added dropwise. The resulting slurry was concentrated under vacuum to about half volume and cooled to 0-5° C. The solid was collected by vacuum filtration, washed with cold water (10 mL), and dried to give 5.3 g (94.9% yield) of 2-amino-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6) δ δ 2.19 (s, 3H), 7.09-7.29 (m, 2H, J=7.5), 7.29-7.43 (d, 1H, J=7.5), 7.61 (s, 2H), 7.85 (s, 1H), 9.63 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ: 18.18, 120.63, 126.84, 127.90, 128.86, 132.41, 133.63, 138.76, 142.88, 159.45, 172.02.
To a stirring solution of compound 5C (5.00 g, 18.67 mmol) and 4,6-dichloro-2-methylpyrimidine (3.65 g 22.4/mmol) in THF (65 mL) was added a 30% wt. solution of sodium t-butoxide in THF (21.1 g, 65.36 mmol) slowly with cooling to keep the temperature at 10-20° C. The mixture was stirred at room temperature for 1.5 h and cooled to 0-5° C. Hydrochloric acid, 2N (21.5 mL) was added slowly and the mixture stirred 1.75 h at 0-5° C. The solid was collected by vacuum filtration, washed with water (15 mL) and dried to give 6.63 g (86.4% yield) of compound 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).
5E. Example 5To a mixture of compound 5D (4.00 g, 10.14 mmol) and hydroxyethylpiperazine (6.60 g, 50.69 mmol) in n-butanol (40 mL) was added DIPEA (3.53 mL, 20.26 mmol). The slurry was heated at 118° C. for 4.5 h, then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with n-butanol (5 mL), and dried. The product (5.11 g) was dissolved in hot 80% EtOH—H2O (80 mL), and the solution was clarified by filtration. The hot solution was slowly diluted with water (15 mL) and cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with 50% ethanol-water (5 mL) and dried affording 4.27 g (83.2% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide as monohydrate. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.40 (s, 3H), 2.42 (t, 2H, J=6), 2.48 (t, 4H, J=6.3), 3.50 (m, 4H), 3.53 (q, 2H, J=6), 4.45 (t, 1H, J=5.3), 6.04 (s, 1H), 7.25 (t, 1H, J=7.6), 7.27 (dd, 1H, J=7.6, 1.7), 7.40 (dd, 1H, J=7.6, 1.7), 8.21 (s, 1H), 9.87 (s, 1H), 11.47.
To a slurry of (E)-N-(2-chloro-6-methylphenyl)-3-ethoxyacrylamide 5B (120 mg, 0.50 mmol) in THF (0.75 ml) and water (0.5 mL) was added NBS (98 mg, 0.55 mmol) at 0° C. The mixture was warmed and stirred at 20-22° C. for 3 h. To this was added 1-(6-chloro-2-methylpyrimidin-4-yl)thiourea 5A (100 mg, 0.49 mmol), and the slurry heated and stirred at reflux for 2 h. The slurry was cooled to 20-22° C. and the solid collected by vacuum filtration giving 140 mg (71% yield) of 2-(6-chloro-2-methylpyrimidin-4-ylamino)-N-(2-chloro-6-methylphenyl)thiazole-5-carboxamide 5D. 1H NMR (400 MHz, DMSO-d6) δ 2.23 (s, 3H), 2.58 (s, 3H), 6.94 (s, 1H), 7.18-7.34, (m, 2H, J=7.5), 7.34-7.46 (d, 1H, J=7.5), 8.31 (s, 1H), 10.02 (s, 1H), 12.25 (s, 1H).Compound 5D was elaborated to N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide, following Step 5E.
2-Piperazin-1-yl-ethanol (8.2 g, 63.1 mmol) was added to a solution of 4,6-dichloro-2-methylpyrimidine (5.2 g, 31.9 mmol) in dichloromethane (80 ml) at rt. The mixture was stirred for two hours and triethylamine (0.9 ml) was added. The mixture was stirred at rt for 20 h. The resultant solid was filtered. The cake was washed with dichloromethane (20 ml). The filtrate was concentrated to give an oil. This oil was dried under high vacuum for 20 h to give a solid. This solid was stirred with heptane (50 ml) at rt for 5 h. Filtration gave 7C (8.13 g) as a white solid
7B. Example 7
To a 250 ml of round bottom flask were charged compound 5C (1.9 g, 7.1 mmol), compound 7C (1.5 g, 5.9 mmol), K2CO3 (16 g, 115.7 mmol), Pd (OAc)2 (52 mg, 0.23 mmol) and BINAP (291 mg, 0.46 mmol). The flask was placed under vacuum and flushed with nitrogen. Toluene was added (60 ml). The suspension was heated to 100-110° C. and stirred at this temperature for 20 h. After cooling to room temperature, the mixture was applied to a silica gel column. The column was first eluted with EtOAC, and then with 10% of MeOH in EtOAC. Finally, the column was washed with 10% 2M ammonia solution in MeOH/90% EtOAC. The fractions which contained the desired product were collected and concentrated to give compound IV as a yellow solid (2.3 g).
Analytical MethodsSolid State Nuclear Magnetic Resonance (SSNMR)All solid-state C-13 NMR measurements were made with a Bruker DSX-400, 400 MHz NMR spectrometer. High resolution spectra were obtained using high-power proton decoupling and the TPPM pulse sequence and ramp amplitude cross-polarization (RAMP-CP) with magic-angle spinning (MAS) at approximately 12 kHz (A. E. Bennett et al, J. Chem. Phys., 1995, 103, 6951), (G. Metz, X. Wu and S. O, Smith, J. Magn. Reson. A, 1994, 110, 219-227). Approximately 70 mg of sample, packed into a canister-design zirconia rotor was used for each experiment. Chemical shifts (6) were referenced to external adamantane with the high frequency resonance being set to 38.56 ppm (W. L. Earl and D. L. VanderHart, J. Magn. Reson., 1982, 48, 35-54).X-Ray Powder DiffractionOne of ordinary skill in the art will appreciate that an X-ray diffraction pattern may be obtained with a measurement error that is dependent upon the measurement conditions employed. In particular, it is generally known that intensities in a X-ray diffraction pattern may fluctuate depending upon measurement conditions employed. It should be further understood that relative intensities may also vary depending upon experimental conditions and, accordingly, the exact order of intensity should not be taken into account. Additionally, a measurement error of diffraction angle for a conventional X-ray diffraction pattern is typically about 5% or less, and such degree of measurement error should be taken into account as pertaining to the aforementioned diffraction angles. Consequently, it is to be understood that the crystal forms of the instant invention are not limited to the crystal forms that provide X-ray diffraction patterns completely identical to the X-ray diffraction patterns depicted in the accompanying Figures disclosed herein. Any crystal forms that provide X-ray diffraction patterns substantially identical to those disclosed in the accompanying Figures fall within the scope of the present invention. The ability to ascertain substantial identities of X-ray diffraction patterns is within the purview of one of ordinary skill in the art.X-Ray powder diffraction data for the crystalline forms of Compound (IV) were obtained using a Bruker GADDS (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) (General Area Detector Diffraction System) manual chi platform goniometer. Powder samples were placed in thin walled glass capillaries of 1 mm or less in diameter; the capillary was rotated during data collection. The sample-detector distance was 17 cm. The radiation was Cu Kα (45 kV 111 mA, λ=1.5418 Å). Data were collected for 3<2θ<35° with a sample exposure time of at least 300 seconds.Single Crystal X-RayAll single crystal data were collected on a Bruker-Nonius (BRUKER AXS, Inc., 5465 East Cheryl Parkway Madison, Wis. 53711 USA) Kappa CCD 2000 system using Cu Kα radiation (λ=1.5418 Å) and were corrected only for the Lorentz-polarization factors. Indexing and processing of the measured intensity data were carried out with the HKL2000 software package (Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W. C. Jr. & Sweet, R. M. (Academic, NY), Vol. 276, pp. 307-326) in the Collect program suite (Data collection and processing user interface: Collect: Data collection software, R. Hooft, Nonius B. V., 1998).The structures were solved by direct methods and refined on the basis of observed reflections using either the SDP (SDP, Structure Determination Package, Enraf-Nonius, Bohemia N.Y. 11716 Scattering factors, including f′ and f″, in the SDP software were taken from the “International Tables for Crystallography”, Kynoch Press, Birmingham, England, 1974; Vol IV, Tables 2.2A and 2.3.1) software package with minor local modifications or the crystallographic package, MAXUS (maXus solution and refinement software suite: S. Mackay, C. J. Gilmore, C. Edwards, M. Tremayne, N. Stewart, K. Shankland. maXus: a computer program for the solution and refinement of crystal structures from diffraction data).The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were varied.The derived atomic parameters (coordinates and temperature factors) were refined through full matrix least-squares. The function minimized in the refinements was Σw(|Fo|−|Fc|)2. R is defined as Σ∥Fo|−|Fc∥/Σ|Fo| while Rw=[Σw(|Fo|−|Fc|)2/Σw|Fo|2]1/2 where w is an appropriate weighting function based on errors in the observed intensities. Difference maps were examined at all stages of refinement. Hydrogens were introduced in idealized positions with isotropic temperature factors, but no hydrogen parameters were variedDifferential Scanning CalorimetryThe DSC instrument used to test the crystalline forms was a TA INSTRUMENTS° model Q1000. The DSC cell/sample chamber was purged with 100 ml/min of ultra-high purity nitrogen gas. The instrument was calibrated with high purity indium. The accuracy of the measured sample temperature with this method is within about +/−1° C., and the heat of fusion can be measured within a relative error of about +/−5%. The sample was placed into an open aluminum DSC pan and measured against an empty reference pan. At least 2 mg of sample powder was placed into the bottom of the pan and lightly tapped down to ensure good contact with the pan. The weight of the sample was measured accurately and recorded to a hundredth of a milligram. The instrument was programmed to heat at 10° C. per minute in the temperature range between 25 and 350° C.The heat flow, which was normalized by a sample weight, was plotted versus the measured sample temperature. The data were reported in units of watts/gram (“W/g”). The plot was made with the endothermic peaks pointing down. The endothermic melt peak was evaluated for extrapolated onset temperature, peak temperature, and heat of fusion in this analysis.Thermogravimetric Analysis (TGA)The TGA instrument used to test the crystalline forms was a TA INSTRUMENTS® model Q500. Samples of at least 10 milligrams were analyzed at a heating rate of 10° C. per minute in the temperature range between 25° C. and about 350° C.
Example 8Preparation of:
Crystalline monohydrate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)An example of the crystallization procedure to obtain the crystalline monohydrate form is shown here:Charge 48 g of the compound of formula (IV).Charge approximately 1056 mL (22 mL/g) of ethyl alcohol, or other suitable alcohol.Charge approximately 144 mL of water.Dissolve the suspension by heating to approximately 75° C.Optional: Polish filter by transfer the compound of formula (IV) solution at 75° C. through the preheated filter and into the receiver.Rinse the dissolution reactor and transfer lines with a mixture of 43 mL of ethanol and 5 mL of water.Heat the contents in the receiver to 75-80° C. and maintain 75-80° C. to achieve complete dissolution.Charge approximately 384 mL of water at a rate such that the batch temperature is maintained between 75-80° C.Cool to 75° C., and, optionally, charge monohydrate seed crystals. Seed crystals are not essential to obtaining monohydrate, but provide better control of the crystallization.Cool to 70° C. and maintain 70° C. for ca. 1 h.Cool from 70 to 5 C over 2 h, and maintain the temperature between 0 at 5° C. for at least 2 h.Filter the crystal slurry.Wash the filter cake with a mixture of 96 mL of ethanol and 96 mL of water.Dry the material at ≦50° C. under reduced pressure until the water content is 3.4 to 4.1% by KF to afford 41 g (85 M %).Alternately, the monohydrate can be obtained by:1) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate and heated at 80° C. to give bulk monohydrate.2) An aqueous solution of the acetate salt of compound IV was seeded with monohydrate. On standing several days at room temperature, bulk monohydrate had formed.3) An aqueous suspension of compound IV was seeded with monohydrate and heated at 70° C. for 4 hours to give bulk monohydrate. In the absence of seeding, an aqueous slurry of compound IV was unchanged after 82 days at room temperature.4) A solution of compound IV in a solvent such as NMP or DMA was treated with water until the solution became cloudy and was held at 75-85° C. for several hours. Monohydrate was isolated after cooling and filtering.5) A solution of compound IV in ethanol, butanol, and water was heated. Seeds of monohydrate were added to the hot solution and then cooled. Monohydrate was isolated upon cooling and filtration.One of ordinary skill in the art will appreciate that the monohydrate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 1 or by a representative sampling of peaks as shown in Table 1.Representative peaks taken from the XRPD of the monohydrate of the compound of formula (IV) are shown in Table 1.TABLE 1 2-Theta d(Å) Height 17.994 4.9257 915 18.440 4.8075 338 19.153 4.6301 644 19.599 4.5258 361 21.252 4.1774 148 24.462 3.6359 250 25.901 3.4371 133 28.052 3.1782 153The XRPD is also characterized by the following list comprising 2θ values selected from the group consisting of: 4.6±0.2, 11.2±0.2, 13.8±0.2, 15.2±0.2, 17.9±0.2, 19.1±0.2, 19.6±0.2, 23.2±0.2, 23.6±0.2. The XRPD is also characterized by the list of 2θ values selected from the group consisting of: 18.0±0.2, 18.4±0.2, 19.2±0.2, 19.6±0.2, 21.2±0.2, 24.5±0.2, 25.9±0.2, and 28.0±0.2.Single crystal x-ray data was obtained at room temperature (+25° C.). The molecular structure was confirmed as a monohydrate form of the compound of Formula (IV).The following unit cell parameters were obtained for the monohydrate of the compound of formula (IV) from the x-ray analysis at 25° C.:a(Å)=13.8632(7); b(Å)=9.3307(3); c(Å)=38.390(2);V(Å3) 4965.9(4); Z′=1; Vm=621Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.354wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).Single crystal x-ray data was also obtained at −50° C. The monohydrate form of the compound of Formula (IV) is characterized by unit cell parameters approximately equal to the following:Cell dimensions: a(Å)=13.862(1);
b(Å)=9.286(1);
c(Å)=38.143(2);
Volume=4910(1) Å3Space group PbcaMolecules/unit cell 8Density (calculated) (g/cm3) 1.369wherein the compound is at a temperature of about −50° C.The simulated XRPD was calculated from the refined atomic parameters at room temperature.The monohydrate of the compound of formula (IV) is represented by the DSC as shown in FIG. 2. The DSC is characterized by a broad peak between approximately 95° C. and 130° C. This peak is broad and variable and corresponds to the loss of one water of hydration as seen in the TGA graph. The DSC also has a characteristic peak at approximately 287° C. which corresponds to the melt of the dehydrated form of the compound of formula (IV).The TGA for the monohydrate of the compound of Formula (IV) is shown in FIG. 2 along with the DSC. The TGA shows a 3.48% weight loss from 50° C. to 175° C. The weight loss corresponds to a loss of one water of hydration from the compound of Formula (IV).The monohydrate may also be prepared by crystallizing from alcoholic solvents, such as methanol, ethanol, propanol, i-propanol, butanol, pentanol, and water.
Example 9Preparation of:
Crystalline n-butanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)The crystalline butanol solvate of the compound of formula (IV) is prepared by dissolving compound (IV) in 1-butanol at reflux (116-118° C.) at a concentration of approximately 1 g/25 mL of solvent. Upon cooling, the butanol solvate crystallizes out of solution. Filter, wash with butanol, and dry.The following unit cell parameters were obtained from the x-ray analysis for the crystalline butanol solvate, obtained at room temperature:a(Å)=22.8102(6); b(Å)=8.4691(3); c(Å)=15.1436(5); β=95.794(2);V(Å3) 2910.5(2); Z′=1; Vm=728Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.283wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the butanol solvate of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 3 or by a representative sampling of peaks. Representative peaks for the crystalline butanol solvate are 2θ values of: 5.9±0.2, 12.0±0.2, 13.0±0.2, 17.7±0.2, 24.1±0.2, and 24.6±0.2.
Example 10Preparation of:
Crystalline ethanol solvate of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV)
To a 100-mL round bottom flask was charged 4.00 g (10.1 mmol) of 5D (contained 2.3 Area % 5C) 6.60 g (50.7 mmol) of 7B, 80 mL of n-butanol and 2.61 g (20.2 mmol) of DIPEA. The resulting slurry was heated to 120° C. and maintained at 120° C. for 4.5 h whereby HPLC analysis showed 0.19 relative Area % of residual 5D to compound IV. The homogeneous mixture was cooled to 20° C. and left stirring overnight. The resulting crystals were filtered. The wet cake was washed twice with 10-mL portions of n-butanol to afford a white crystalline product. HPLC analysis showed this material to contain 99.7 Area % compound IV and 0.3 Area % 5C.The resulting wet cake was returned to the 100-mL reactor, and charged with 56 mL (12 mL/g) of 200 proof ethanol. At 80° C. an additional 25 mL of ethanol was added. To this mixture was added 10 mL of water resulting in rapid dissolution. Heat was removed and crystallization was observed at 75-77° C. The crystal slurry was further cooled to 20° C. and filtered. The wet cake was washed once with 10 mL of 1:1 ethanol:water and once with 10 mL of n-heptane. The wet cake contained 1.0% water by KF and 8.10% volatiles by LOD. The material was dried at 60° C./30 in Hg for 17 h to afford 3.55 g (70 M %) of material containing only 0.19% water by KF, 99.87 Area % by HPLC. The 1H NMR spectrum, however revealed that the ethanol solvate had been formed.The following unit cell parameters were obtained from the x-ray analysis for the crystalline ethanol solvate (di-ethanolate, E2-1), obtained at −40° C.:a(Å)=22.076(1); b(Å)=8.9612(2); c(Å)=16.8764(3); β=114.783(1);V(Å3) 3031.1(1); Z′=1; Vm=758Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.271wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 4 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 5.8±0.2, 11.3±0.2, 15.8±0.2, 17.2±0.2, 19.5±0.2, 24.1±0.2, 25.3±0.2, and 26.2±0.2.In addition, during the process to form the ethanolate (diethanolate) the formation of another ethanol solvate (½ ethanolate, T1E2-1) has been observed. To date this additional ethanol solvate is known strictly as a partial desolvation product of the original diethanolate form E2-1, and has only been observed on occasion during crystallization of E2-1The following unit cell parameters were obtained from the x-ray analysis for the crystalline ½ ethanol solvate T1E2-1, obtained at −10° C.:a(Å)=22.03(2); b(Å)=9.20(1); c(Å)=12.31(1);β=93.49(6)V(Å3) 2491(4)); Z′=1; Vm=623;Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.363wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the ethanol solvate (T1E2-1) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 7 or by a representative sampling of peaks. Representative peaks for the crystalline ethanol solvate are 2θ values of: 7.20±0.2, 12.01±0.2, 12.81±0.2, 18.06±0.2, 19.30±0.2, and 25.24±0.2.
Example 11Preparation of:
Crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (Neat form N-6)To a mixture of compound 5D (175.45 g, 0.445 mol) and hydroxyethylpiperazine (289.67 g, 2.225 mol) in NMP (1168 mL) was added DIPEA (155 mL, 0.89 mol). The suspension was heated at 110° C. (solution obtained) for 25 min., then cooled to about 90° C. The resulting hot solution was added dropwise into hot (80° C.) water (8010) mL, keeping the temperature at about 80° C. The resulting suspension was stirred 15 min at 80° C. then cooled slowly to room temperature. The solid was collected by vacuum filtration, washed with water (2×1600 mL) and dried in vacuo at 55-60° C. affording 192.45 g (88.7% yield) of N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide. 1H NMR (400 MHz, DMSO-d6): δ 2.24 (s, 3H), 2.41 (s, 3H), 2.43 (t, 2H, J=6), 2.49 (t, 4H, J=6.3), 3.51 (m, 4H), 3.54 (q, 2H, J=6), 4.46 (t, 1H, J=5.3), 6.05 (s, 1H), 7.26 (t, 1H, J=7.6), 7.28 (dd, 1H, J=7.6, 1.7), 7.41 (dd, 1H, J=7.6, 1.7), 8.23 (s, 1H), 9.89 (s, 1H), 11.48. KF0.84; DSC: 285.25° C. (onset), 286.28° C. (max).The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline compound IV, obtained at 23° C.:a(Å)=22.957(1); b(Å)=8.5830(5); c(Å)=13.803(3); β=112.039(6);V(Å3)=2521.0(5); Z′=1; Vm=630Space group P21/aMolecules/unit cell 4Density (calculated) (g/cm3) 1.286wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the crystalline form of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 5 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (N-6) are 2θ values of: 6.8±0.2, 11.1±0.2, 12.3±0.2, 13.2±0.2, 13.7±0.2, 16.7±0.2, 21.0±0.2, 24.3±0.2, and 24.8±0.2.
Example 12Preparation of:
Crystalline N-(2-chloro-6-methylphenyl)-2-(6-(4-(3-hydroxyethyl)piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (IV) (neat form T1H1-7)The title neat form may be prepared by heating the monohydrate form of the compound of formula (IV) above the dehydration temperature.The following unit cell parameters were obtained from the x-ray analysis for the neat crystalline (T1H1-7) compound IV, obtained at 25° C.:a(Å)=13.4916; b(Å)=9.3992(2); c(Å)=38.817(1);V(Å3)=4922.4(3); Z′=1; Vm=615Space group PbcaDensity (calculated) (g/cm3) 1.317wherein Z′=number of drug molecules per asymmetric unit. Vm=V(unit cell)/(Z drug molecules per cell).One of ordinary skill in the art will appreciate that the neat crystalline form (T1H1-7) of the compound of formula (IV) may be represented by the XRPD as shown in FIG. 6 or by a representative sampling of peaks. Representative peaks for the crystalline neat form (T1H1-7)) are 2θ values of: 8.0±0.2, 9.7±0.2, 11.2±0.2, 13.3±0.2, 17.5±0.2, 18.9±0.2, 21.0±0.2, 22.0±0.2.Obviously, numerous modifications and variations of the present invention are possible in light of the above teachings. It is therefore to be understood that within the scope of the appended claims, the invention may be practiced otherwise than as specifically described herein. PAPERhttps://pubs.acs.org/doi/abs/10.1021/jm060727j
2-Aminothiazole (1) was discovered as a novel Src family kinase inhibitor template through screening of our internal compound collection. Optimization through successive structure−activity relationship iterations identified analogs 2 (Dasatinib, BMS-354825) and 12m as pan-Src inhibitors with nanomolar to subnanomolar potencies in biochemical and cellular assays. Molecular modeling was used to construct a putative binding model for Lck inhibition by this class of compounds. The framework of key hydrogen-bond interactions proposed by this model was in agreement with the subsequent, published crystal structure of 2 bound to structurally similar Abl kinase. The oral efficacy of this class of inhibitors was demonstrated with 12m in inhibiting the proinflammatory cytokine IL-2 ex vivo in mice (ED50 ∼ 5 mg/kg) and in reducing TNF levels in an acute murine model of inflammation (90% inhibition in LPS-induced TNFα production when dosed orally at 60 mg/kg, 2 h prior to LPS administration). The oral efficacy of 12m was further demonstrated in a chronic model of adjuvant arthritis in rats with established disease when administered orally at 0.3 and 3 mg/kg twice daily. Dasatinib (2) is currently in clinical trials for the treatment of chronic myelogenous leukemia.
PATENTSPublication numberPriority datePublication dateAssigneeTitleUS20060079563A1 *1999-04-152006-04-13Jagabandhu DasCyclic protein tyrosine kinase inhibitorsUS20070219370A1 *2006-03-152007-09-20Bristol-Myers Squibb CompanyProcess for preparing n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino] -5-thiazolecarboxamide and related metabolites thereofUS20080275009A1 *2005-09-212008-11-06Bristol-Myers Squibb CompanyOral administration of n-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-1,3-thiazole-5-carboxamide and salts thereofUS20090030203A1 *2005-08-052009-01-29Bristol-Myers Squibb CompanyPreparation of 2-amino-thiazole-5-carboxylic-acid derivativesUS20090118297A1 *2007-10-232009-05-07Ondrej SimoPolymorphs of dasatinib and process for preparation thereofWO2012014149A12010-07-302012-02-02Ranbaxy Laboratories LimitedN-methylformamide solvate of dasatinibUS20120309968A1 *2010-02-082012-12-06Nan Jing Cavendish Bio-Engineering Technology Co., Ltd.Polymorphs of dasatinib, preparation methods and pharmaceutical compositions thereofUS8530492B22009-04-172013-09-10Nektar TherapeuticsOligomer-protein tyrosine kinase inhibitor conjugatesUS8680103B22004-02-062014-03-25Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-aromatic carboxamides as kinase inhibitorsWO2014102759A22012-12-312014-07-03Ranbaxy Laboratories LimitedProcess for the preparation of dasatinib and its intermediatesUS8816077B22009-04-172014-08-26Nektar TherapeuticsOligomer-protein tyrosine kinase inhibitor conjugatesUS20150057446A1 *2012-04-202015-02-26Shilpa Medicare LimitedProcess for preparing dasatinib monohydrateWO2016001025A12014-06-302016-01-07Basf SeMulticomponent crystals of dasatinib with menthol or vanillinUS9340536B22012-06-152016-05-17Basf SeMulticomponent crystals comprising dasatinib and selected co-crystal formersUS9556164B22013-07-252017-01-31Basf SeSalts of Dasatinib in crystalline formUS9884857B22013-07-252018-02-06Basf SeSalts of dasatinib in amorphous formWO2018078392A12016-10-292018-05-03Cipla LimitedPolymorphs of dasatinibWO2018100585A12016-12-012018-06-07Natco Pharma LimitedAn improved process for the preparation of dasatinib polymorphWO2018134189A12017-01-202018-07-26Cerbios-Pharma SaCo-crystal of an antitumoral compoundWO2018134190A12017-01-202018-07-26Cerbios-Pharma SaCo-crystals of an antitumoral compoundUS10174018B22016-12-132019-01-08Princeton Drug Discovery IncProtein kinase inhibitorsWO2019209908A12018-04-252019-10-31Johnson Matthey Public Limited CompanyCrystalline forms of dasatinibUS10722484B22016-03-092020-07-28K-Gen, Inc.Methods of cancer treatmentUS10799459B12019-05-172020-10-13Xspray Microparticles AbRapidly disintegrating solid oral dosage forms containing dasatinibFamily To Family CitationsUS7396935B22003-05-012008-07-08Bristol-Myers Squibb CompanyAryl-substituted pyrazole-amide compounds useful as kinase inhibitorsUS7652146B2 *2004-02-062010-01-26Bristol-Myers Squibb CompanyProcess for preparing 2-aminothiazole-5-carboxamides useful as kinase inhibitorsTW200600513A *2004-06-302006-01-01Squibb Bristol Myers CoA method for preparing pyrrolotriazine compoundsPE20061394A1 *2005-03-152006-12-15Squibb Bristol Myers CoMetabolites of n- (2-chloro-6-methylphenyl) -2 – [[6- [4- (2-hydroxyethyl) -1-piperazinyl] -2-methyl-4-pyrimidinyl] amino] -5-thiazolecarboxamidesUS20060235006A1 *2005-04-132006-10-19Lee Francis YCombinations, methods and compositions for treating cancerPL1885339T32005-05-052015-12-31Bristol Myers Squibb Holdings IrelandFormulations of a src/abl inhibitorWO2008076883A22006-12-152008-06-26Abraxis Bioscience, Inc.Triazine derivatives and their therapeutical applicationsWO2010139979A22009-06-032010-12-09Generics [Uk] LimitedProcesses for preparing crystalline formsWO2010139980A1 *2009-06-032010-12-09Generics [Uk] LimitedProcess for preparing crystalline dasatinib monohydrateEP2359813A12010-02-042011-08-24Ratiopharm GmbHPharmaceutical composition comprising N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamidCN102250084A *2010-02-082011-11-23南京卡文迪许生物工程技术有限公司Dasatinib polymorphic substance as well as preparation method and pharmaceutical composition thereofCN102643275B *2011-02-212016-04-20江苏先声药物研究有限公司The preparation method that a kind of Dasatinib N-6 crystal formation is newWO2013065063A12011-11-032013-05-10Cadila Healthcare LimitedAnhydrous form of dasatinib, process for its preparation and its useUS20150087687A12012-03-232015-03-26Dennis BrownCompositions and methods to improve the therapeutic benefit of indirubin and analogs thereof, including meisoindigoSG10201610869TA2012-06-262017-02-27Del Mar PharmaceuticalsMethods for treating tyrosine-kinase-inhibitor-resistant malignancies in patients with genetic polymorphisms or ahi1 dysregulations or mutations employing dianhydrogalactitol, diacetyldianhydrogalactiCN103664929B *2012-08-302016-08-03石药集团中奇制药技术(石家庄)有限公司Dasatinib polycrystalline form medicament and preparation methodCN102838595B *2012-09-132014-09-24江苏奥赛康药业股份有限公司Preparation method of high-purity dasatinib and by-product of dasatinibCN103819469A *2012-11-162014-05-28重庆医药工业研究院有限责任公司Crystal form of dasatinib and preparation method for crystal form of dasatinibCZ306598B62012-12-062017-03-22Zentiva, K.S.A method of preparation and purification of new and known polymorphs and dasatinib solvatesCN105764502A2013-07-262016-07-13现代化制药公司Combinatorial methods to improve the therapeutic benefit of bisantrene and analogs and derivatives thereofCN103408542B *2013-08-132016-06-29南京优科生物医药研究有限公司A kind of preparation method of highly purified Dasatinib anhydrideWO2015049645A2 *2013-10-042015-04-09Alembic Pharmaceuticals LimitedAn improved process for the preparation of dasatinibCZ306732B62013-12-192017-05-31Zentiva, K.S.A method of preparation of the anhydrous polymorphic form of N-6 DasatinibCN104788445B *2015-04-102017-06-23山东新时代药业有限公司A kind of synthetic method of Dasatinib intermediateCN106668022B *2015-11-052020-09-15武汉应内药业有限公司Application of aminothiazole MyD88 specific inhibitor TJM2010-5* Cited by examiner, † Cited by third party, ‡ Family to family citation
^World Health Organization (2019). World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. hdl:10665/325771. WHO/MVP/EMP/IAU/2019.06. License: CC BY-NC-SA 3.0 IGO.
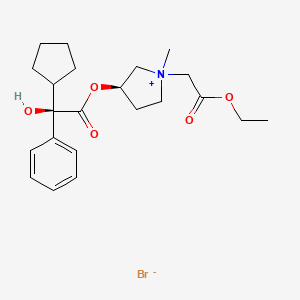
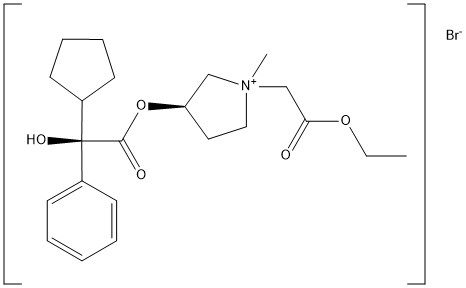
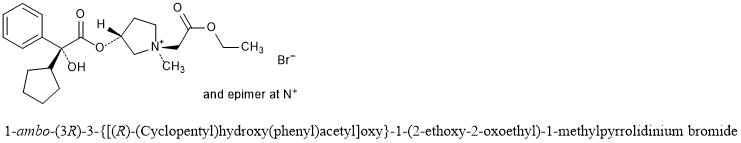






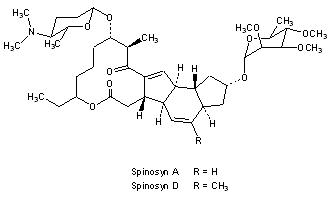



 Figure 5.10 Chemical synthesis steps in spinetoram manufacturing.
Figure 5.10 Chemical synthesis steps in spinetoram manufacturing.
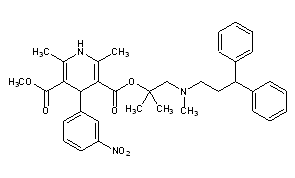
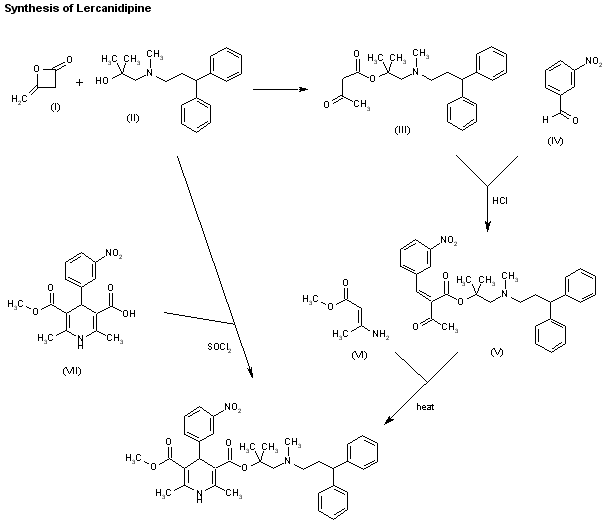







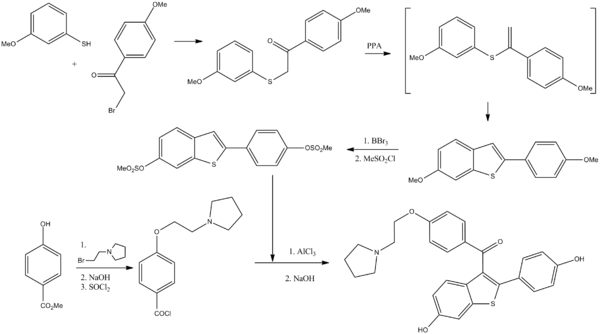
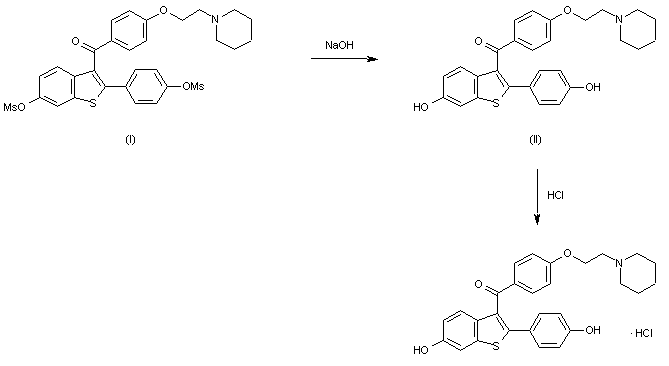

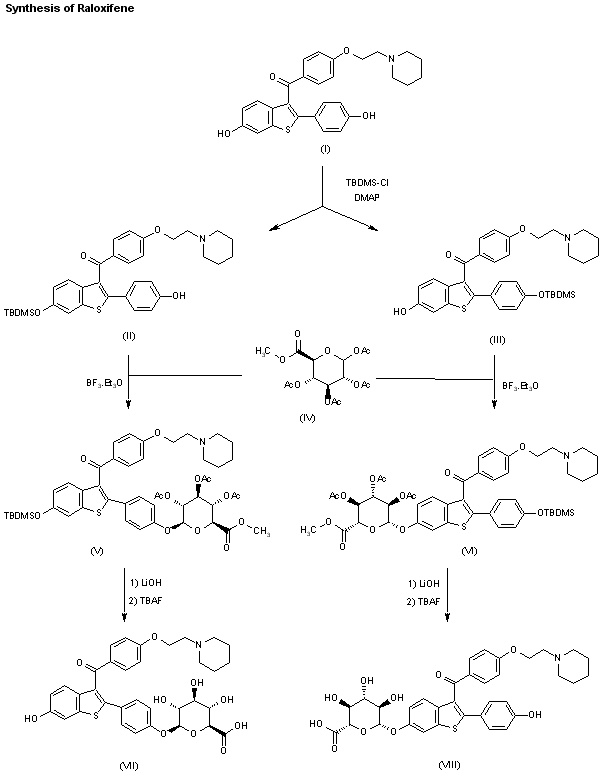
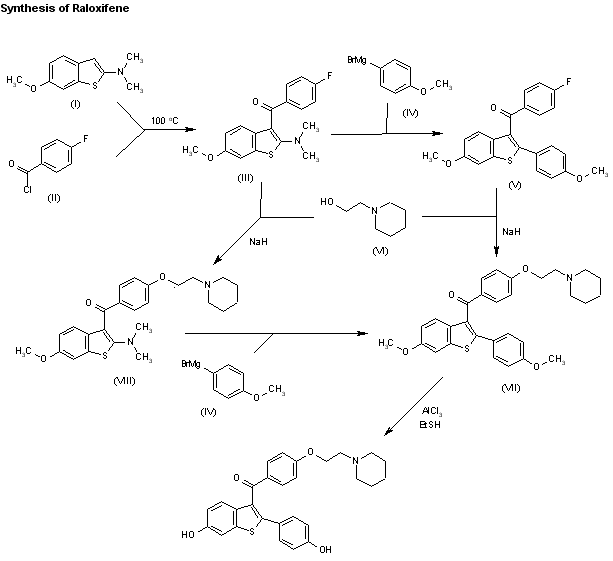














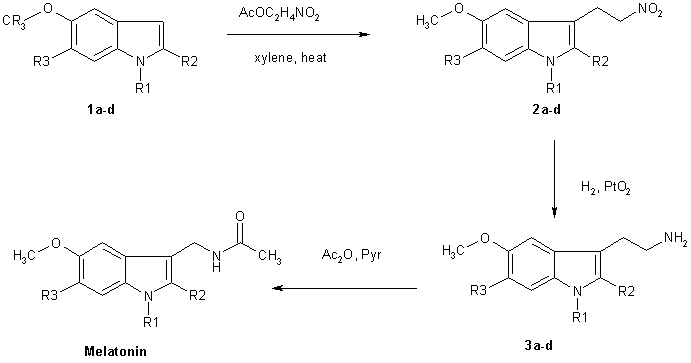
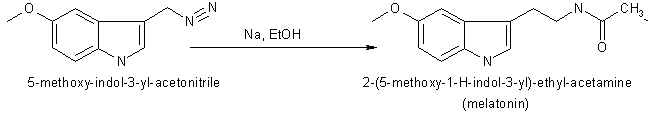
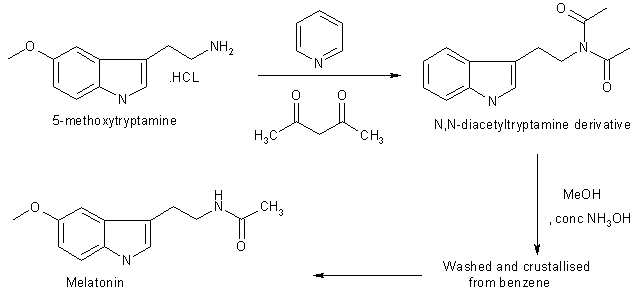
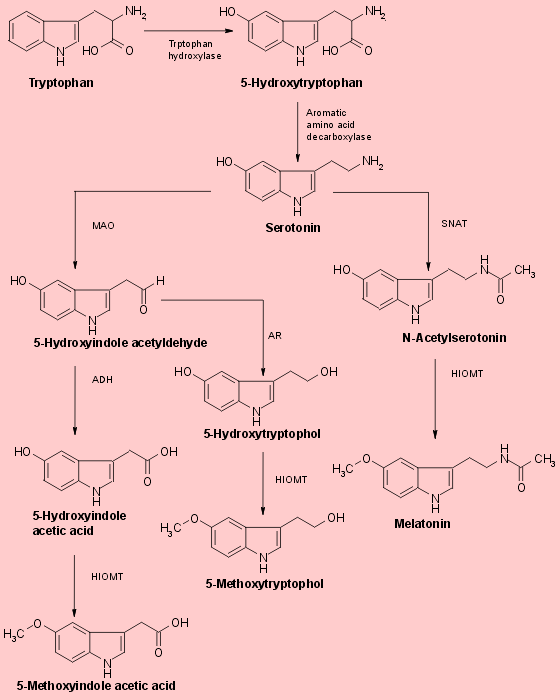 Fig.1 Biosynthesis of pineal methoxyindoles from serotonin
Fig.1 Biosynthesis of pineal methoxyindoles from serotonin



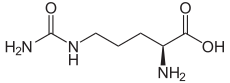



 ). A constant stream of argon gas is passed through the solution to remove the residual sulfide as hydrogen disulfide. The pH of the solution is then raised to 5.9 ± 0.2 using sodium hydroxide to form isoelectric L-citrulline.
). A constant stream of argon gas is passed through the solution to remove the residual sulfide as hydrogen disulfide. The pH of the solution is then raised to 5.9 ± 0.2 using sodium hydroxide to form isoelectric L-citrulline.