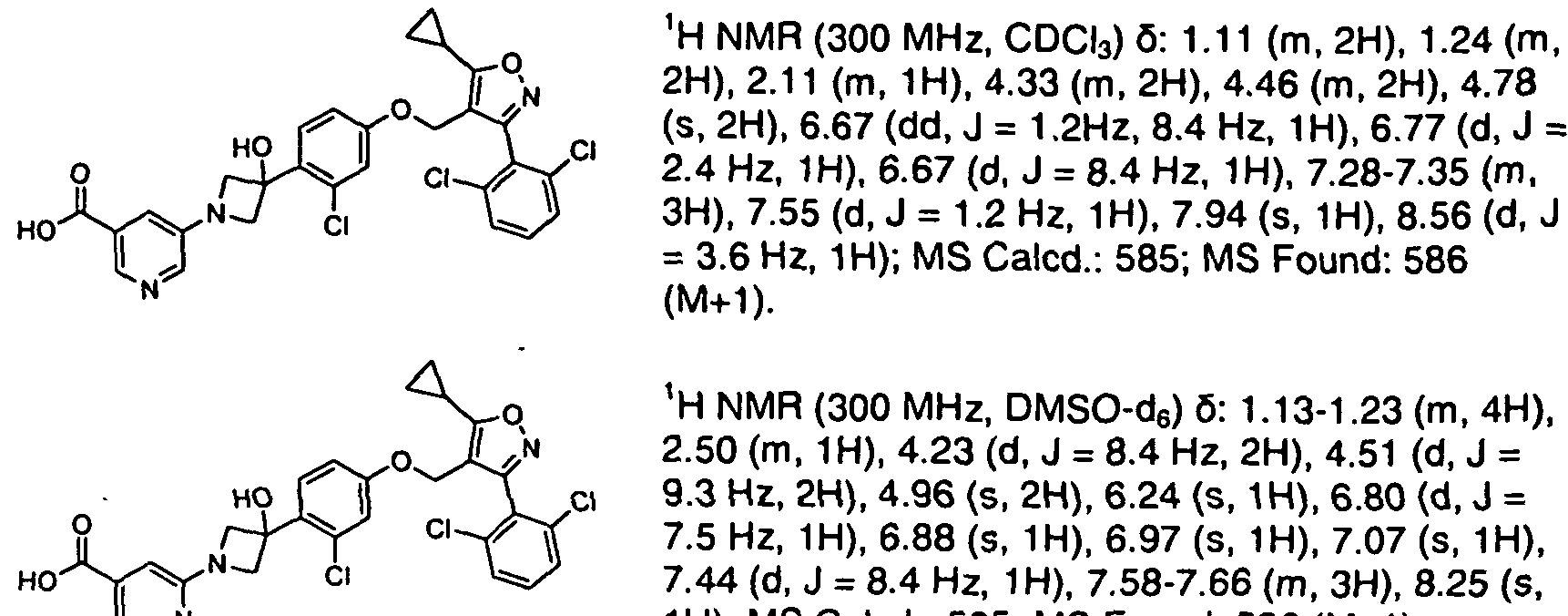![Kuvan (Saproterin Dihydrochloride Tablets): Uses, Dosage, Side Effects, Interactions, Warning]()
Sapropterin
Sapropterin dihydrochloride, Dapropterin dihydrochloride, R-THBP, 6R-BH4, SUN-0588, Phenoptin, Biopten, Biobuden, Bipten
Approval:US: Dec’07, EU: Dec’08
Approval:US: Dec’07, EU: Dec’08
IUPAC Name
(6R)-2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-3,4,5,6,7,8-hexahydropteridin-4-one
SMILES
[H][C@@]1(CNC2=C(N1)C(=O)NC(N)=N2)[C@@H](O)[C@H](C)O
сапроптерин [Russian] [INN]
- 17528-72-2
- 27070-47-9
- Sun 0588
- 6R-BH4
- R-THBP
- Sapropterin
- Sapropterina
- sapropterinum
- Tetrahydrobiopterin
Title: Sapropterin
CAS Registry Number: 62989-33-7
CAS Name: (6R)-2-Amino-6-[(1R,2S)-1,2-dihydroxypropyl]-5,6,7,8-tetrahydro-4(1H)-pteridinone
Additional Names: (6R)-L-erythro-tetrahydrobiopterin; dapropterin; R-THBP; 6R-BH4
Molecular Formula: C9H15N5O3
Molecular Weight: 241.25
Percent Composition: C 44.81%, H 6.27%, N 29.03%, O 19.90%
Literature References: Natural cofactor of the aromatic amino acid hydroxylases required for catecholamine and serotonin biosynthesis. Identification of cofactor activity: S. Kaufman, Proc. Natl. Acad. Sci. USA 50, 1085 (1963). Prepn of (6R,S)-BH4: B. Schircks et al., Helv. Chim. Acta 61, 2731 (1978). Chromatographic separation of diastereoisomers: S. W. Bailey, J. E. Ayling, J. Biol. Chem. 253, 1598 (1978). Absolute configuration of natural isomer: W. L. F. Armarego et al., Aust. J. Chem. 35, 785 (1982). Stereospecific synthesis: S. Matsuura et al., Heterocycles 23, 3115 (1985); H. Sakai, T. Kanai, EP 191335; eidem, US 4713454 (1986, 1987 both to Shiratori; Suntory). Bioavailability: G. Kapatos, S. Kaufman, Science 212, 955 (1981). Effect on neurotransmitter monoamine biosynthesis: S. Miwa et al., Arch. Biochem. Biophys. 239, 234 (1985). LC determn in biological samples: Y. Tani, T. Ishihara, Life Sci. 46, 373 (1990). Therapeutic potential in hyperphenylalaninemia: S. Kaufman, J. Nutr. Sci. Vitaminol, Suppl., 601 (1992).
Properties: pK¢ 5.05. uv max (0.1 N HCl): 265 nm (e 14000).
pKa: pK¢ 5.05
Absorption maximum: uv max (0.1 N HCl): 265 nm (e 14000)
Derivative Type: Dihydrochloride
CAS Registry Number: 69056-38-8
Manufacturers’ Codes: SUN-0588
Trademarks: Biopten (Maruho)
Molecular Formula: C9H15N5O3.2HCl
Molecular Weight: 314.17
Percent Composition: C 34.41%, H 5.45%, N 22.29%, O 15.28%, Cl 22.57%
Properties: Crystals from HCl, mp 245-246° (dec). [a]D25 -6.81° (c = 0.665 in 0.1 M HCl). uv max (2 M HCl): 264 nm (e 16770).
Melting point: mp 245-246° (dec)
Optical Rotation: [a]D25 -6.81° (c = 0.665 in 0.1 M HCl)
Absorption maximum: uv max (2 M HCl): 264 nm (e 16770)
Therap-Cat: In treatment of hyperphenylalaninemia.
Keywords: Enzyme Cofactor
Experimental Properties
| PROPERTY |
VALUE |
SOURCE |
| melting point (°C) |
250-255 °C (hydrochloride salt) |
Not Available |
| water solubility |
>20 mg/mL (dichloride salt) |
Not Available |
| logP |
-1.7 |
Not Available |
Tetrahydrobiopterin (BH4, THB), also known as sapropterin (INN),[2][3] is a cofactor of the three aromatic amino acid hydroxylase enzymes,[4] used in the degradation of amino acid phenylalanine and in the biosynthesis of the neurotransmitters serotonin (5-hydroxytryptamine, 5-HT), melatonin, dopamine, norepinephrine (noradrenaline), epinephrine (adrenaline), and is a cofactor for the production of nitric oxide (NO) by the nitric oxide syntheses.[5] Chemically, its structure is that of a (dihydropteridine reductase) reduced pteridine derivative (Quinonoid dihydrobiopterin).[6]
Medical use
Tetrahydrobiopterin is available as a tablet for oral administration in the form of sapropterin dihydrochloride (BH4*2HCL).[7][8][9] It was approved for use in the United States as a tablet in December 2007[10][11] and as a powder in December 2013.[12][11] It was approved for use in the European Union in December 2008,[9] Canada in April 2010,[11] and Japan in July 2008.[11] It is sold under the brand names Kuvan and Biopten.[9][8][11] The typical cost of treating a patient with Kuvan is US$100,000 per year.[13] BioMarin holds the patent for Kuvan until at least 2024, but Par Pharmaceutical has a right to produce a generic version by 2020.[14]
Sapropterin is indicated in tetrahydrobiopterin deficiency caused by GTP cyclohydrolase I (GTPCH) deficiency, or 6-pyruvoyltetrahydropterin synthase (PTPS) deficiency.[15] Also, BH4*2HCL is FDA approved for use in phenylketonuria (PKU), along with dietary measures.[16] However, most people with PKU have little or no benefit from BH4*2HCL.[17]
Sapropterin (tetrahydrobiopterin or BH4) is a cofactor in the synthesis of nitric oxide. It is also essential in the conversion of phenylalanine to tyrosine by the enzyme phenylalanine-4-hydroxylase; the conversion of tyrosine to L-dopa by the enzyme tyrosine hydroxylase; and conversion of tryptophan to 5-hydroxytryptophan via tryptophan hydroxylase.
Sapropterin commonly known as tetrahydrobiopterin (THB or BH4) developed by BioMarin and marketed as Sapropterin dihydrochloride under the brand name of KUVAN®. It is indicated for the treatment of phenylketonuria (PKU) and tetrahydrobiopterin deficiencies. Sapropterin dihydrochloride is chemically known as (6R)-2-amino-6-[(lR, 2S)-1, 2- dihydroxypropyl]-5,6,7,8-tetrahydro-4(lH)-pteridinone dihydrochloride and structurally represented as below.
Sapropterin dihydrochloride
Due to its vital role in the conversion of L-tyrosine into L-DOPA, which is the precursor for dopamine, a deficiency in tetrahydrobiopterin can cause severe neurological disorders unrelated to toxic build-up of L-phenylalanine; dopamine is a crucial neurotransmitter, and is the precursor of norepinephrine and epinephrine. Thus, a deficiency of tetrahydrobiopterin can result in phenylketonuria (PKU) from L-phenylalanine concentrations or hyperphenylalaninemia (HP A), as well as monoamine and nitric oxide neurotransmitter deficiency or chemical imbalance. The chronic presence of PKU can result in severe brain damage, including symptoms of mental retardation, speech impediments like stuttering, slurring, seizures or convulsions and behavioural abnormalities.
In an article published in Bio Chem J 347 (1): 1-16, tetrahydrobiopterin is reported to be biosynthesized from guanosine triphosphate (GTP) by three chemical reactions mediated by the enzymes GTP cyclohydrolase I (GTPCH), 6-pyruvoyltetrahydropterin synthase (PTPS), and sepiapterin reductase (SR).
Preparation of Sapropterin is reported with a mixture of R & S isomers in Helv. Chim. Acta, 60, 1977, 211-214, by catalytic reduction of L-biopterin of formula (2). Similar process with slight modifications is also published in Hel. Chim. Acta, 61, 1978, 2731- 2738.
(2)
In another publication reported in Helv. Chim. Acta, 62, 1979, 2577-2580, separation of the diastereomers (6R) and (6S)-5,6,7,8-tetrahydro-L-biopterin is reported by fractional crystallization of corresponding tetraacetyl derivative followed by hydrolysis using aq. HC1.
In another process published in Heterocycles, 23(12), 1985, 3115-3120, Sapropterin dihydrochloride of formula (1) is prepared by catalytic hydrogenation of L- biopterin of formula (2) in the presence of Pt02 under latm hydrogen pressure in 0.1 M potassium phosphate buffer at pH 11.8 for 18hr followed by filtration and recrystallization from 8M HC1. With slight modifications in the above reaction conditions like using platinum black, aq. base solutions like tetraethylammonium hydroxide or triethylamine etc. under 100 Kg/cm2 hydrogen pressure / 0° C / pH 12.0 / 1000 rpm / 20h/3N HCl-EtOH with 85% yield is disclosed in US4713454. In another process disclosed in US4595752, L-biopterin of formula (2) is catalytically reduced in the presence of platinum oxide in aq. base / acid solutions like (10% aq. potassium carbonate, aq. sodium carbonate, aq. potassium acetate and 0.1 N aq. HCl) under bubbling of hydrogen gas for 5-30hr at room temperature followed by filtration and isolated as HCl salt of formula (1) using aq. HCl and ethanol to obtain Sapropterin dihydrochloride.
In another approach disclosed in WO2005049614, racemic isomers of Sapropterin dihydrochloride are prepared from L-neopterin.
In another process disclosed in WO2009088979, the diacetyl biopterin is hydrolysed in the presence of aq. diethyl amine-n-butanol mixture at 40°C for 16hr at pH >11.5 followed by hydrogenation in the presence of platinum black using 50 bar hydrogen pressure at 25 °C. Product of formula (1) isolated as HCl salt from ethanol or butanol.
In another process disclosed in US20130197222, Sapropterin dihydrochloride of formula (1) is prepared starting from condensation of crotonoic acid.
The process for preparation of key intermediate, L-biopterin of formula (2) is cited in the following references.
In an article published in J. Am. Chem. Soc, 1955, 77, 3167-3168, L-biopterin of formula (2) is reported to be first isolated from human urine. The melting point reported to be 250-280°C. In another article published in J. Am. Chem. Soc, 1956, 78, 5868-5871, L-biopterin of formula (2) is prepared starting from L-rhamnose. A slight modification in the reaction conditions mentioned above is disclosed in US3505329.
In the article published in Helv. Chim. Acta, 1969, 52, 1225-1228, L-biopterin of formula (2) along with 7-biopterin is synthesized by condensing 2, 4, 5-triamino-6-oxo-l, 6-dihydropyrimidine dihydrochloride with (1 -benzyl- l-phenyl-hydrazino)-5-desoxy-L- ribulose followed by oxidation of the tetrahydro derivative.
Later in the year 1974, in an article, J. Am. Chem. Soc, 1974, 96, 6781-6782, L-biopterin is reported to be prepared starting from L-rhamnose. In another approach published in Bull. Chem. Soc. Jpn., 1975, 48(12), 3767-3768, L- biopterin of formula (2) is prepared from 2, 4, 5-triamino-6-hydroxypyrimidine dihydrochloride is reacted with hydrazone derivative in aq. methanol at reflux temperature.
In another process disclosed in US5043446 (1989), L-biopterin process is claimed to be synthesized starting from D-ribose. Similar approach with slight variations in the process, later published in Liebigs Ann. Chem., 1989, 1267-1269.
In another approach published in Agric. Biol. Chem., 1989, 53, 2095-2100, L-biopterin is synthesized starting from (S)-ethyl lactate. Prior to this publication the methodology is claimed by the same authors in JP01-221380 (1989).
In another approach disclosed in US5037981 (1990), L-biopterin is synthesized from 2- methylfuran.
In the article, Synthesis, 1992, 303-308, L-biopterin is synthesized from (4S)-4(3P- Acetoxy-5-androsten-17P-ylcarbonyloxy)-2-pentynol.
In the approach published in J. Org. Chem., 1996, 61, 8698-8700, L-biopterin is synthesized from L-tartaric acid.
In the patent US7361759 (2005), L-biopterin of formula (2) is made from L-rhamnose diethyl mercaptal.
US 20120157671 application discloses the preparation of compound of formula (4a) is by reacting D-ribose of formula (3) with acetone in the presence of sulphuric acid at room temperature followed by neutralization with sodium carbonate and concentrated under vacuum.
![Sapropterin | Nature Reviews Drug Discovery]()
![Pharmaceutics 12 00323 g004 550]()
https://www.mdpi.com/1999-4923/12/4/323/htm
Synthesis Reference
Steven S. Gross, “Blocking utilization of tetrahydrobiopterin to block induction of nitric oxide synthesis.” U.S. Patent US5502050, issued October, 1984.
US5502050
SYN
![]()
SYN
![]()
![]()
![]()
![]()
Synthetic Reference
Hong, Hao; Gage, James; Chen, Chaoyong; Lu, Jiangping; Zhou, Yan; Liu, Shuangyong. Method for synthesizing sapropterin dihydrochloride. Assignee Asymchem Laboratories (Tianjin) Co., Ltd., Peop. Rep. China; Asymchem Life Science (Tianjin) Co., Ltd.; Tianjin Asymchem Pharmaceutical Co., Ltd.; Asymchem Laboratories (Fuxin) Co., Ltd.; Jilin Asymchem Laboratories Co., Ltd. WO 2013152609. (2013).
syn 1
EP 0191335. Aust J Chem 1984,37(2),355-66, Chem Lett 1984,5(5),735-8
Helv Chim Acta 1979,62(8),2577-80
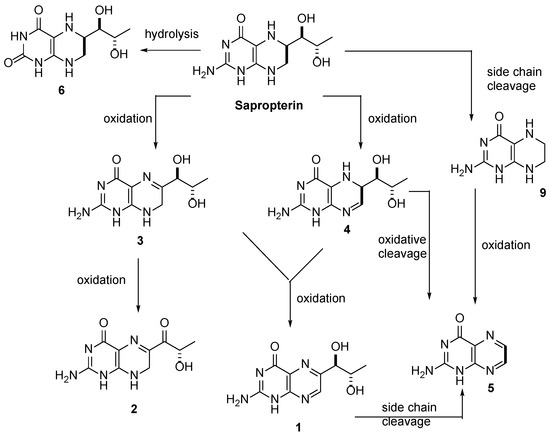
This compound can be prepared in two related ways: 1) The catalytic hydrogenation of biopterin (I) with H2 over PtO2 aqueous K2HPO4 at pH 11.4 or aq. (Et)4NOH at pH 12 yields a solution which is acidified with HCl. After evaporation, the residue is crystallized in ethanol – HCl. 2) The acetylation of biopterin (I) with refluxing acetic anhydride gives the triacetyl derivative (II), which is hydrogenated with H2 over PtO2 in trifluoroacetic acid, yielding the (6RS)-mixture of triacetyl derivatives (III). Acetylation of (III) with refluxing acetic anhydride affords the tetracetyl (6RS)-derivative (IV), which by fractional crystallization or column chromatography of the dihydrochloride in methanol gives the desired compound as pure (6R)-isomer.
![]()
PATENT
https://patents.google.com/patent/WO2016189542A1/en
formula 1).
The present invention is shown in below scheme- 1
Experimental Section: Example-1: Preparation of (6R)-2-amino-6-[(lR, 2S)-1, 2-dihydroxypropyl]-5,6,7,8- tetrahydro-4(lH)-pteridinone dihydrochloride of formula (1):
Step (i): Preparation of 2, 3-O-isopropylidene-D-ribose of formula (4a)
Into a 5L, 4 necked round-bottomed flask equipped with a mechanical stirrer, a thermometer socket, and a condenser, were charged acetone (3.0 L), D-ribose (300.0 gm, 2.0 mole) and p-toluene sulfonic acid (11.5 gm). The solution was stirred and maintained at 20-25°C for 2.5-3.0hrs. After completion of reaction, the reaction mixture was neutralized with aq. base solution and filtered. The filtrate was evaporated to dryness to get 375.0 gm (98.8% by theory) of 2, 3-O-isopropylidene-D-ribose of formula (4a) as light brown colour oily residue. Purity: >95% by GC. Step (ii): Preparation of l-deoxy-3, 4-O-isopropylidene-D-allitol of formula (5a)
Into a 5L, 4 necked round-bottomed flask equipped with a mechanical stirrer, a thermometer socket, and a condenser, was charged, 2.0L, 3M methyl magnesium chloride and cooled to 10° C. To this stirred solution, a solution of 200gm of 2,3-0- isopropylidene-D-ribose of formula (4a) dissolved in 200 mL tetrahydrofuran was added. After completion of reaction, the reaction mixture was quenched with ammonium chloride, extracted with ethyl acetate and separated. The solvent was evaporated to dryness under vacuum to get 185gm of l-deoxy-3, 4-O-isopropylidene-D-allitol of formula (5a) as dark brown colour oily residue. The crude product was purified by crystallization from ethyl acetate/hexane mixture to get 130g (60% by theory) as white crystalline solid. Purity: >98% by GC.
JR (λ Cm-1, KBr disc): 3317.64, 2993.69-2976.90, 2926.08, 2873.26 (m) -CH3, 1074.35; 1 HNMR (400 MHz, DMSO-d6, EDl®j&¾ : (H2¾H3, J=6.8Hz, 3H),
1.148 (s, CH3, 3H), 1.290 (s,CH3), 3.415-3.357 (m, CH, 1H), 3.652-3.571 (m, CH2, 2H), 3.812-3.803 (d, 2 X CH, 2H), 4.00-3.969 (q, CH, 1H), 4.504-4.476 (t, ΟΗ, ΙΗ), 4.504- 4.476 (d, OH, 1H), 5.381-5.371 (d, OH, 1H): 13 CNMR (100 MHz, DMSO-d6, □ (ppm): 20.59, 25.35, 27.73, 63.18, 64.61 , 69.77, 76.82, 81.40, 107.31 ; Mass: 206.42 [M], 205.41 [M-l]. DSC (° C): 77.58° C Step (iii): Preparation of 5-deoxy-2, 3-O-isopropylidene-D-ribose of formula (6a)
Into a 5L 4 necked round-bottomed flask equipped with a mechanical stirrer, a thermometer socket, and a condenser, were charged, 1.6 L of water and 270 gm of sodium meta periodate. The solution was cooled to 10-20°C. To the stirred solution, a solution of 200 gm of l-deoxy-3, 4-O-isopropylidene-D-allitol of formula (5a) dissolved in 1.4 L of isopropyl ether at 25°C. After addition, the reaction mixture was maintained at 25-30° C for l-2h. After completion of reaction, the layers were separated and the organic layer was washed with water, aq. sodium bicarbonate and separated. The excess solvent was removed by distillation under vacuum to get 145 gm (85.4% by theory) of 5- deoxy-2, 3-O-isopropylidene-D-ribose of formula (6a) as yellow oil. Purity: >98% by GC.
Step (iv & v): Preparation of 5-deoxy-L-ribose phenyl hydrazone of formula (8) a) Step (iv): Preparation of 5-deoxy-L-ribose of formula (7)
Into a 2L 4 necked round-bottomed flask equipped with a mechanical stirrer, a thermometer socket, and a condenser, were charged 600ml of water and 200gm of 5- deoxy-2, 3-O-isopropylidene-D-ribose of formula (6a). To the stirred reaction mixture, 180gm of resin was charged and stirred for 8-10 h at 10-15° C. After completion of reaction, the resin was recovered and the filtrate was clarified by activated charcoal and filtered. The filtrate was distilled off under vacuum and the resulting 5-deoxy-L-ribose of formula (7) present water was directly used in the next step without further isolation and purification. The purity of 5-deoxy-L-ribose of formula (7) present in water was above 95% by TLC.
b) Step (v): Preparation of 5-deoxy-L-ribose phenyl hydrazone of formula (8)
Into a 2L 4 necked round-bottomed flask equipped with a mechanical stirrer, a thermometer socket, and a condenser, were charged the above aq. solution of 5-deoxy-L- ribose of formula (7), 5.0 mL of acetic acid. To the stirred solution, 125g of phenyl hydrazine was charged and stirred the reaction mixture for l-2h at 25-35° C. After completion of reaction, the reaction product was filtered and washed with isopropyl ether. The wet product was dried to get 190g (73.9% by theory) of 5-deoxy-L-ribose phenyl hydrazone of formula (8) as yellow colour crystalline powder. Purity: >99.0% by HPLC. Step (VI toX): Preparation of L-erythro-biopterin of formula (2)
a) Step (vi): Preparation of triacetoxy-5-deoxy-L-ribose phenylhydrazone of formula (9)
Into a 10L 4 necked round-bottomed flask equipped with a mechanical stirrer, a thermometer socket, and a guard tube, were charged 5L of ethyl acetate, 500g of 5- deoxy-L-ribose phenyl hydrazone of formula (8) and 54gm of 4-dimethylaminopyridine. The reaction mixture was cooled to 25-30° C and was added 730gm of acetic anhydride drop wise. The reaction mixture was maintained under stirring for 2-3h. After completion of reaction, the reaction mixture was washed with water, aq. sodium carbonate and water, and separated. The organic layer was used in the next stage without further isolation and purification.
b) Step (vii): Preparation of 1,2-diacetyl-biopterin of formula (10)
Into a 20L 4 necked round-bottomed flask equipped with a mechanical stirrer, a thermometer socket, addition funnel, and a condenser, were charged, the above organic layer containing triacetoxy-5-deoxy-phenyl hydrazone of formula (9) obtained in step (vi), 3.0 L methanol and 4-hydroxy-2,5,6-triaminopyrimidine base (generated from 600 gm of corresponding sulphate salt) and salt (generated from 350 gm of tetra butyl ammonium bromide and 154g of 70% perchloric acid) and 5.3L water under stirring and heated and maintained at 35-40°C for 6-8h. The reaction mixture was then cooled to 20- 25°C and added 1.0 Kg 35% aq. hydrogen peroxide drop wise. The reaction mixture was maintained for 36-40h under stirring at 25-30°C and resulting product was filtered under suction. The wet product was washed with water and utilized in the next step without further purification.
c) Step (viii): Preparation oi -erythro biopterin of formula (2)
Into a 10L 4-necked round-bottomed flask equipped with a mechanical stirrer, condenser, thermometer socket, and addition funnel, were charged 1.35 L of aq. potassium hydroxide and the above wet product obtained from step (vii). The reaction mixture was heated to 45-50° C and maintained form 2-3h and filtered. The pH of the filtrate was adjusted to neutral and the resulting product was filtered and dried to get 205 g of crude L-erythro-biopterin of formula (2) as dark brown solid. Purity: >90% by HPLC
d) Step (ix): Preparation of potassium salt oi -erythro biopterin of formula (11a) Into a 10L 4 necked round-bottomed flask equipped with a mechanical stirrer, thermometer socket, and a glass stopper, were charged 650 mL water followed by HOg of potassium hydroxide and dissolved under stirring. The potassium hydroxide solution was cooled to 25-30° C and the above crude L-erythro-biopterin of formula (2) was charged under stirring. The resulting solution was then clarified using activated carbon and filtered. The potassium salt was regenerated from the solution by the addition of 8.5L of isopropyl alcohol. The resulting salt was filtered and washed with isopropyl alcohol. The wet product of formula (11a) was utilized in the next step without further purification.
e) Step (x): Preparation of pure L-er thro biopterin of formula (2) from potassium salt of L-erythro biopterin of formula (2)
Into a 5L 4 necked round-bottomed flask equipped with mechanical stirrer, thermometer socket, and addition funnel, were charged 3.2 L of water and the above wet potassium salt of formula (11a). The reaction mixture was stirred to dissolve completely. The resulting solution was clarified using activated carbon and filtered. The pH of the filtrate was adjusted to 6.0-7.0 to get pure L-erythro-biopterin of formula (2). The product was filtered and washed with water followed by isopropyl alcohol followed by isopropyl ether to get 130g of highly pure L-erythro biopterin of formula (2) with > 98% HPLC purity Appearance: pale brown coloured solid.
1H NMR (3N DC1) 5(ppm): 1.569-1.585(d, 3H), 4.596-4.657(p, 1H), 5.325-5.337(d, 1H), 9.355(s, 1H); Mass: 238.29(M+1), 239.22(M+2).
Step (xi): Preparation of Sapropterin dihydrochloride of formula (1)
Into a 5L 4 necked round-bottomed flask equipped with mechanical stirrer, and thermometer socket, were charged 1.8L of water, 250g of L-erythro-biopterin of formula (2) followed by 800mL of 20% aq. potassium carbonate solution under stirring. The solution was then added 90g of platinum oxide catalyst. The reaction mixture was then transferred into an autoclave and pressurized with 40 bar hydrogen gas and hydrogenated at room temperature for 24-30h under stirring. After completion of reaction, the catalyst was filtered off and the pH of the filtrate was acidified with concentrated hydrochloric acid. The water was evaporated under vacuum and the resulting crude Sapropterin dihydrochloride of formula (1) was isolated as pale yellow colour solid by addition of isopropanol/l-pentanol mixture. The product was dried in a vacuum oven to get 250g of crude Sapropterin dihydrochloride of formula (1). Step (xii): Purification of Sapropterin dihydrochloride of formula (1)
Into a 2L 4 necked round-bottomed flask equipped with a mechanical stirrer, thermometer socket, and reflux condenser, were charged 1L water and 250g of Sapropterin dihydrochloride of formula (1). The contents were stirred to dissolve completely. The clear solution was treated with activated charcoal and filtered. The filtrate was distilled off completely under vacuum to afford pale yellow solid. The product was isolated from isopropanol/l-pentanol mixture to get 225.0 g (90%) pure Sapropterin dihydrochloride of formula (1) as pale yellow to off-white solid. HPLC purity is >99.9%.
Example 2: Preparation of triacetoxy-5-deoxy-L-ribose phenylhydrazone of formula
(9)
Into a 10L 4 necked round-bottomed flask equipped with a mechanical stirrer, a thermometer socket, and a guard tube, were charged 50mL of ethyl acetate, 5.0g of 5- deoxy-L-ribose phenyl hydrazone of formula (8) and 0.54g of N, N-dimethylamino pyridine. The reaction mixture was cooled to 15-20°C and was added 7.2gm of acetic anhydride drop wise. The reaction mixture was maintained under stirring for 6-8h. After completion of reaction, the reaction mixture was washed with water, aq. sodium carbonate and water, and separated. The organic layer was distilled under reduced pressure and product was isolated from n-hexane to get 6.2g of triacetoxy-5 -deoxy-L- ribose phenylhydrazone of formula (9) 79.4% yield.
Appearance: Orange coloured solid.
Melting point: 70-75 °C.
1HNMR (CDC13): 1.275-1.29 l(d, 3H), 2.039(s, 3H), 2.085-2.095(d, 6H), 5.083-5.144(m, 1H), 5.390-5.416(t, 1H), 5.589-5.619(t, 1H), 6.849-6.886(t, 1H), 6.922-6.937(t, 1H), 6.966-6.987(d, 2H), 7.221-7.242(d, 2H), 7.563(s, 1H(D20 exchangeable).
13CNMR (CDC13): 15.325, 20.816-21.053, 68.482, 71.717, 73.043, 112.759, 120.510, 129.212, 132.105, 144.049, 169.496, 169.948. Example 3: Preparation of potassium salt of L-erythro biopterin of formula (11)
Into a 1.0L 4 necked round-bottomed flask equipped with a mechanical stirrer, thermometer socket, and a glass stopper, were charged 75 mL water followed by 3.7g of potassium hydroxide and dissolved under stirring. The potassium hydroxide solution was cooled to 25-30° C and 15.0g of crude L-erythro-biopterin of formula (2) was charged under stirring. The resulting solution was then clarified using activated carbon and filtered. The potassium salt was regenerated from the solution by the addition of 500mL of ethanol. The resulting salt was filtered and washed with ethanol and dried to get 9.1g of potassium salt of L-erythro biopterin of formula (11) with 52.3% yield. HPLC <98% Appearance: Brown coloured solid.
1H NMR (D20): 1.187-1.203(d, 3H), 4.158-4.220(p, 1H), 4.731-4.745(d, 1H), 8.623(s, 1H).
13C NMR (D20): 18.198, 70.645, 76.703, 128.811, 147.875, 149.410, 156.504, 164.774, 173.731.
Mass: 276.23(M+1), 277.21(M+2), 238.29(M-K+1); DSC (° C): 313.12°
Example 4: Preparation of Sapropterin dihydrochloride of formula (1)
Into a 5L 4 necked round-bottomed flask equipped with mechanical stirrer, and thermometer socket, were charged 1.8L of water, 250g of L-erythro-biopterin of formula (2) followed by 800ml of 20% aq. potassium hydroxide solution under stirring. The solution was then added 90gm of platinum oxide catalyst. The reaction mixture was then transferred into an autoclave and pressurized with 50 bar hydrogen gas and hydrogenated at room temperature for 24-30h under stirring. After completion of reaction, the catalyst was recovered by filtration and the filtrate was acidified with concentrated hydrochloric acid. The water was evaporated under vacuum and the resulting crude Sapropterin dihydrochloride of formula (1) was isolated as pale yellow colour solid by addition of ethanol- 1 -pentanol mixture. The product was dried in a vacuum oven to get 250g of crude Sapropterin dihydrochloride of formula (1). Example 5: Purification of Sapropterin dihydrochloride of formula (1)
Into a 2L 4 necked round-bottomed flask equipped with a mechanical stirrer, thermometer socket, and reflux condenser, were charged 1L water and 250g of Sapropterin dihydrochloride of formula (1). The contents were stirred to dissolve completely and the clear solution was treated with activated charcoal and filtered. The filtrate was distilled off completely under vacuum to afford pale yellow solid. The product 225.0 g (90%) was isolated ethanol- 1 -pentanol mixture as pure Sapropterin dihydrochloride of formula (1) as pale yellow to off-white solid. HPLC purity is >99.9%.
syn
![str1]()
![str2]()
![str1]()
![str2]()
![str3]()
PATENT
https://patents.google.com/patent/WO2016101211A1/en
was developed by Merck and was launched in the United States and the European Union in 2007 and 2008 under the trade name Kuvan. This product can be used to treat hyperphenylalaninemia (HPA) caused by tetrahydrobiopterin (BH4) deficiency. The structure is as follows:
The chemical name is: (6R)-2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-5,6,7,8-tetrahydro-4(1H)-fluorenone Dihydrochloride.
The oxaprozin hydrochloride can be obtained by hydrogenation of L-erythrobiopterin. The literature Liebigs Ann. Chem. 1989, 1267-1269 reports the preparation of L-erythrobiopterin starting from L-ribose. The preparation route is as follows:
Although the method is simple and easy to perform, it is a better preparation route, but the disadvantage is that the starting material L-ribose price is higher, thus causing the cost of sapropium hydrochloride to be high.
The literature for the preparation of L-erythrobiopteris is reported by the documents Helv. Chim. Acta, 1985, 1639-1643, US2011218339A, etc. The product of the acetylation reaction of the steroid compound 6 with 2,4,5-triaminopyrimidinone Cyclization in a methanol/water/pyridine system followed by aromatization with an iodine reagent to give an acetylated L- Red-type biopterin, followed by hydrolysis and deacetylation to obtain L-erythrobiopterin. The reaction equation is as follows:
![Figure PCTCN2014094961-appb-000003]()
Among them, compound 6 is used as a key intermediate, and many methods for its preparation are reported. The method reported in J. Am. Chem. Soc. 1974, 6781-6782, J. Am. Chem. Soc. 1976, 2301-2307, etc., uses L-rhamnose as a raw material, and reacts with ethanethiol to form a corresponding shrinkage. Sulfuraldehyde, oxidizing thiol to sulfone with an oxidizing agent, removing a carbon under alkaline conditions to obtain 5-deoxy-L-arabinose, and reacting 5-deoxy-L-arabinose with phenylhydrazine to obtain a key intermediate formula 6 . The synthetic route is as follows:
Although this method has been improved and improved many times, the ethanethiol used has a special malodor and requires the use of a deodorizing device, and its lower boiling point also causes inconvenience to the production.
Document J. Org. Chem. 1996, 8699-8700 reports that L-tartaric acid is used as a starting material, which is protected by hydroxyl group, carboxyl group, reduction, addition, deprotection to obtain 5-deoxy-L-ribose, 5-deoxy- The condensation of L-arabinose with phenylhydrazine gives key intermediates. The synthetic route is as follows:
The reducing agent used in the route of the acid chloride to reduce the aldehyde is bis(triphenylphosphine) copper borohydride (I), which has a high price and is not favorable for the control of industrialization cost. The reaction temperature of the format reagent with carbonyl addition and lactone reduction is -78 ° C, and the energy consumption in industrial production is high. In addition, the post-treatment of the multi-step reaction uses silica gel column color The spectrum is purified and it is difficult to achieve industrialization. Therefore, this route has great disadvantages in terms of cost and operability in industrial production.
Document CN201010151443.2 reports the use of L-arabinose as a starting material to obtain L-erythrobioptery through a multi-step reaction. The preparation route is as follows:
In reproducing the preparation method, we have found that the intermediate 2 is directly subjected to reduction and desulfonation reaction to prepare the intermediate 2, which has the disadvantages of low yield, low product purity, and difficulty in purification of the product. Therefore, it is necessary to find a simple, feasible and low-cost preparation route.
scheme synthetic route includes the following steps:
Example 1: Preparation of Product 1
To the reaction flask was added 10 L of anhydrous methanol, and 1.5 kg of the starting material L-arabinose was added under mechanical stirring. 250 g of concentrated sulfuric acid was added dropwise under a water bath, and the reaction was stirred for 20-24 hours. The reaction was monitored by TLC, and 350 g of sodium carbonate was added to the reaction system. Stir until pH = 7-8 and filter. The filtrate was concentrated under reduced pressure at 35 ° C to 40 ° C to dryness to yield 1.64 kg of oil, yield -100%.
Example 2: Preparation of product 2
The product 1, 4 L of pyridine and 5 L of acetonitrile were added to the reaction flask and dissolved by mechanical stirring. The mixture was cooled by stirring, and a solution of 1.8 kg of p-toluenesulfonyl 5 L acetonitrile was added dropwise at a temperature of 0 to 5 ° C. After completion of the dropwise addition, the reaction was stirred at room temperature 20-25 ° C for 4 hours. The TLC monitors the reaction.
After concentration, 12 L of ethyl acetate and 5 L of water were added to the concentrated residue, and the layers were stirred. The organic layer was washed with 1 mol/L hydrochloric acid, saturated sodium hydrogen carbonate and saturated brine and dried. Filtration and concentration of the filtrate gave 1.7 kg of pale yellow oil, yield 56.3%.
Example 3: Preparation of product 3
1.2 kg of product 2 was added to a 10 L reaction flask, dissolved with 6 L of methyl ethyl ketone, and 840 g of sodium iodide was added with stirring. After the addition, the temperature was refluxed for 12 hours, and the reaction was completed by TLC. The mixture was cooled to room temperature, filtered, and the filtrate was evaporated. It was dissolved in ethyl acetate, washed with water, and the aqueous layer was evaporated. The combined organic layers were washed with EtOAc EtOAc m.
Example 4: Preparation of product 4
To a 20 L reaction flask was added 900 g of product 3, 332 g of triethylamine dissolved in 9 L of methanol, 45 g of 10% Pd/C, vacuumed, hydrogenated twice, and hydrogenated at a constant temperature of 25-30 ° C for 16 hours. The reaction was completed by TLC, filtered, and the filtrate was concentrated under reduced pressure to give a residue. 4 L of ethyl acetate was added to the residue to precipitate a white solid. The mixture was stirred at 0 ° C for 30 min, and filtered. The filtrate was added to 2 L of a 0.4 mol/L sulfuric acid solution and the layers were separated. The aqueous layer was washed once with 50 mL of ethyl acetate to give an aqueous solution of product 4 (approximately 250 g).
Example 5: Preparation of product 5
The aqueous solution of product 4 was added to the reaction flask, and the reaction was heated at 75 ° C for 3 hours, and the reaction was completed by TLC (DCM: MeOH = 10:1). After cooling to room temperature, it was washed with 100 mL of ethyl acetate, and the aqueous layer was separated to give the product 5, i.e., about 213 g of aqueous solution of 5-deoxy-L-arabinose, which was directly reacted in the next step.
Example 6: Preparation of product 6
To the reaction flask, 2.5 L of ethyl acetate and 170 g of phenylhydrazine were added under nitrogen, and an aqueous solution of the product 5 was added dropwise with stirring at a temperature of 5 to 10 ° C (protected from light). The reaction was kept for 1 hour, and then the temperature was raised to 20-25 ° C for 30 min. The reaction was completed by TLC and the layers were separated. The aqueous layer was extracted with ethyl acetate and organic layers were combined. The organic layer was dried over anhydrous sodium sulfate and filtered.
The ethyl acetate solution of product 6 was added to the reaction flask under nitrogen, and 8 L of petroleum ether was slowly added with stirring. After the addition was completed, the mixture was cooled to -5 – 10 ° C and stirred for 1 hour, and filtered to give a beige solid. Drying under reduced pressure at 30-35 ° C gave a dry product of about 250 g, yield 71.4%.
Example 7: Preparation of product 7
To the reaction flask was added 2.5 L of ethyl acetate and 250 g of product 6. 30 g of DMAP was added with stirring. 400 ml of acetic anhydride was added dropwise at a temperature of 15 ° C, and the reaction was stirred at a temperature of 20-25 ° C for 3 hours. The reaction was monitored by TLC, and a hydrochloric acid solution was added at a temperature of 15 ° C to separate the layers. The organic layer was washed with saturated hydrochloric acid and saturated sodium hydrogen sulfate. The organic phase was separated, dried and filtered to give 371 g, m.
Example 8: Preparation of product 9
To the reaction flask was added 220 g of product 8, 2.2 L of purified water. Under stirring, 500 g of a product 7 in 5 L of methanol and 150 g of anhydrous lithium perchlorate dissolved in 1.5 L of water were added. After the addition was completed, the reaction was stirred at a temperature of 30 to 32 ° C for 20 hours. The reaction is completed and filtered. The filtrate was temperature-controlled at 15 ° C to 20 ° C, and 1 L of 30% hydrogen peroxide was added dropwise. After the addition, the reaction was kept at 20 ° C for 6 hours, and the solid was precipitated, filtered, and dried by blasting at 35-40 ° C to obtain 215 g of a brownish yellow product 9 in a yield of 47%.
Example 9: Preparation of product 10
To the reaction flask, 80 g of product 9, 400 ml of purified water, 300 ml of n-butanol, and 80 ml of diethylamine were added, and the mixture was stirred and heated to 45-50 ° C for 16 hours. After the TLC reaction is completed, the layers are separated, and the aqueous layer is separated to obtain an aqueous solution of the product 10, which is directly reacted in the next step.
Example 10: Preparation of Product I
An aqueous solution of product 10 was added to the autoclave, and 50 ml of triethylamine and 2 g of platinum dioxide were added thereto with stirring. The pressure was evacuated, the hydrogen was replaced three times, the pressure was controlled to 1.5 MPa, and the reaction was stirred at 35 ° C for 20 hours. After filtration, the filtrate was added to 30 ml of n-butanol for 5 min, and the mixture was allowed to stand to give an aqueous solution of product I. 200 ml of concentrated hydrochloric acid was added dropwise at a temperature of 10 ° C, and the aqueous solution was concentrated under reduced pressure to dryness. 500 ml of 95% ethanol was added to the crude product, and the mixture was heated to 55-60 ° C for 1 hour, then cooled to 35 ° C for 2 hours, filtered, and the filter cake was dried to give the product I35 g.
Example 11: Preparation of product 9′
To the reaction flask was added 1.25 L of ethyl acetate and 125 g of product 9. 15 g of DMAP was added with stirring. 200 ml of acetic anhydride was added dropwise at a temperature of 15 ° C, and the reaction was stirred at a temperature of 20-25 ° C for 3 hours. The reaction was monitored by TLC, and a hydrochloric acid solution was added at a temperature of 15 ° C to separate the layers. The organic layer was washed with saturated hydrochloric acid and saturated sodium hydrogen sulfate. The organic phase was separated, dried and concentrated to give 12,5 g of oil.
Example 12: Preparation of product 10
The product 9′ prepared in Example 11 was added to the reaction flask, 600 ml of purified water, 450 ml of n-butanol, and 120 ml of diethylamine were added, and the mixture was stirred and heated to 45-50 ° C for 16 hours. After the TLC reaction is completed, the layers are separated, and the aqueous layer is separated to obtain an aqueous solution of the product 10, which is directly reacted in the next step.
Example 13: Preparation of Product I
An aqueous solution of the product 10 prepared in Example 12 was added to the hydrogenation vessel, and 80 ml of triethylamine, 3 g of platinum dioxide was added thereto with stirring, and vacuum was applied thereto, and the pressure was controlled to 1.5 MPa, and the reaction was stirred at 35 ° C for 20 hours. After filtration, the filtrate was added to 45 ml of n-butanol for 5 min, and the mixture was allowed to stand to give an aqueous solution of product I. After cooling at 10 ° C, 300 ml of concentrated hydrochloric acid was added dropwise, and the aqueous solution was concentrated under reduced pressure to dryness. 750 ml of 95% ethanol was added to the crude product, and the mixture was heated to 55-60 ° C for 1 hour, then cooled to 35 ° C for 2 hours, filtered, and the filter cake was dried to give the product I 48.9 g.
///////////
https://patents.google.com/patent/US9365573B2/en
Sapropterin dihydrochloride, chemical name (6R)-2-amino-6-[(1R,2S)-1,2-dihydroxypropyl]-5,6,7,8-tetrahydro-4(1H)-pteridinone dihydrochloride, molecular formula C9H15N5O3.2HCl, and CAS registry number 69056-38-28, is a synthetic product of tetrahydrobiopterin (BH4) dihydrochloride. BH4 is a cofactor of Phenylalanine Hydroxylase (PAH). Tyrosine is acquired from Phenylalanine (Phe) through hydroxylation under the action of PAH which is low in activity or even inactive in PKU patients, while BH4 is able to activate PAH, promote normal oxidative metabolism of Phe in the bodies of the patients, and reduce the Phe levels in the bodies of some patients. On Dec. 16, 2007, the sapropterin dihydrochloride tablets produced by BioMarin Pharmaceutical Inc. in USA were approved by the Food and Drug Administration (FDA) for marketing for treatment of PKU. Because of the effective activity of sapropterin dihydrochloride, it is extremely necessary to select a route applicable to industrial production with high product purity.
At present, BH4 is mainly synthesized by the following methods reported in literatures:
1. Preparation using 4-hydroxy-2,5,6-triaminopyrimidine (TAP) and 5-deoxy-L-arabinose as raw materials, please see literature E. L. Patterson et al., J. Am. Chem. Soc. 78, 5868(1956).
2. Preparation using TAP and 5-deoxy-L-arabinose phenylhydrazone as raw materials, please see literature Matsuura et al., Bull. Chem. Soc. Jpn., 48,3767 (1975);
3. Preparation by reaction of raw materials hydroxyl-protected TAP and 4-acetyl-2,3-epoxypentanal through oxidation of iodine and a dehydroxylation protecting group, please see literature Matsuura et al., Chemistry of Organic Synthesis, MI/g. 46. No. 6, P570(1988).
These traditional methods for preparing BH4 have the following major disadvantages: raw materials are expensive, arabinose which can be hardly acquired is used as a carbon atom radical for asymmetric synthesis; there are multiple steps in reactions with low yield, and low product purity, 5-deoxy-L-arabinose is easily degraded in a reaction solution, and products of the synthesis routes above have low stereoselectivity. To sum up, the traditional synthesis methods are not applicable to mass industrial production. Therefore, a synthesis route, which is applicable to industrial production with high product purity, high yield and high stereoselectivity, needs to be searched urgently.
tep 10: add 0.7 kg (0.05 g/g) of palladium 5% on carbon in the presence of the methanol solution containing 1.5 kg of acetylamino-7,8-dihydropteridine
obtained in Step 9, introduce hydrogen until the pressure of the reaction kettle is 0.8±0.05 MPa, control the temperature of the system at 25±5° C. and the pressure at 0.8±0.05 MPa, react for 82 hours, after reacting thoroughly, perform quenching in 31.9 kg (9 eq) of dilute hydrochloric acid having a concentration of 15%, and perform suction filtration and drying to the system to obtain a target product, i.e. a crude product of sapropterin dihydrochloride
recrystallize and purify the crude product by 29 L (20 ml/g) of methanol at 35±5° C. to obtain 0.8 kg of a pure product, with a yield of 45%, a purity of 98.3% and an enantiomeric excess of 99.1%.
Embodiment 5: main raw material:
and X═O
Step 1: add 836 kg (0.3 eq) of a tetrahydrofuran solution contaning a samarium catalyst having a concentration of 4%, 29.2 kg (0.3 eq) of (R)-(+)-1,1′-bi-2-naphthol, 28.4 kg (0.3 eq) of triphenylphosphine oxide, and 600 kg (10 kg/kg) of a 4 A molecular sieve to a 3000 L reaction kettle, after stirring uniformly, control the system temperature at 20±5° C., add 117.4 kg (2 eq) of meta-chloroperoxybenzoic acid, add 60 kg (1 eq) of benzyl crotonate
to the system after adding meta-chloroperoxybenzoic acid, react for 32 hours while preserving the temperature, add 19.6 kg (0.3 eq) of citric acid to the system to stop the reaction, and perform centrifugation, concentration and rectification to the system to obtain 40.5 kg of (2S,3R)-2,3-epoxy-benzyl butyrate
with a yield of 62%;
Step 2: add 36.8 kg (3 eq) of acetone, and 5.4 kg (0.6 eq) of lithium chloride to a 500 L enamel vessel, control the temperature at 15±5° C., add 40.5 kg (1 eq) of (2S,3R)-2,3-epoxy-benzyl butyrate
react for 7 hours while preserving the temperature, add 422 kg (2 eq) of a potassium bicarbonate aqueous solution having a concentration of 10%, and perform liquid separation, extraction and concentration to the system to obtain 44 kg of (4S,5S)-2,2,5-trimethyl-acetonide-benzyl butyrate
with a yield of 82%;
Step 3: add 352 L (8 ml/g) of ethanol, and 44 kg (1 eq) of (4S,5S)-2,2,5-trimethyl-2,3-acetonide-benzyl butyrate
to a 1000 L reaction kettle, increase the temperature to 37±5° C., add 4.8 kg (1.5 eq) of pure water and 53.2 kg (1.5 eq) of a sodium hydroxide aqueous solution having a concentration of 20%, react for 6 hours while preserving the temperature, perform centrifugation, dissolve a filter cake in 352 L (8 ml/g) of ethanol, add 71.0 kg (3 eq) of L-α-amphetamine, preserve the temperature at 22±5° C. for 4 hours, and perform centrifugation and drying to obtain 32.4 kg of (4S,5S)-2,2,5-trimethyl-2,3-acetonide-phenylacetylamino butyrate
with a yield of 62%;
Step 4: add 48 L (6 ml/g) of 1,4-dioxane, 8 kg (1 eq) of (4S,5S)-2,2,5-trimethyl-2,3-acetonide-phenylacetylamino butyrate
to a 72 L reaction bottle, then add a dilute sulphuric acid aqueous solution having a concentration of 10% to the system to regulate the pH at 2.5±0.5, control the temperature at −5±5° C., react for 1 hour, perform liquid separation to obtain an organic phase, add 7.0 kg of (2.0 eq) N,N-diisopropylethylamine to the organic phase, and concentrate the system to obtain 4.1 kg of (4S,5S)-2,2,5-trimethyl-1,3-dioxolan-4-methanoic acid
with a yield of 93.5%;
Step 5: add 49 L (12 ml/g) of 2-methyltetrahydrofuran, 4.1 kg of 1,3-dioxolan-4-methanoic acid
and 13.1 kg (4 eq) of N,N-diisopropylethylamine to a 100 L reaction bottle, reduce the temperature to −22±5° C., add 5.5 kg (2.0 eq) of ethyl chloroformate, react for 1.8 hours while preserving the temperature, introduce a diazomethane gas for 1.8 hours, add 18.5 kg (4.5 eq) of a hydrochloride ethanol solution having a concentration of 20%, react for 1.8 hours, add potassium bicarbonate to regulate the pH value to 8.5±0.5, and perform extraction, liquid separation and concentration to obtain 4.1 kg of (4S,5S)-2,2,5-trimethyl-5-chloroacetyl-1,3-dioxolane
with a yield of 83.7%;
Step 6: add 49 L (12 ml/g) of acetone, 4.1 kg of (4S,5S)-2,2,5-trimethyl-5-chloroacetyl-1,3-dioxolane
3.4 kg (2.5 eq) of sodium azide, and 1.8 kg (0.5 eq) of potassium iodide to a 72 L bottle, react the system for 26 hours while preserving the temperature at 34±5° C., perform filtering and concentration to obtain an acetone solution containing 3.9 kg of (4S,5S)-2,2,5-trimethyl-5-(2-azidoacetyl)-1,3-dioxolane
with a yield of 91.5%;
Step 7: add 46.4 L (12 ml/g) of methyl tert-butyl ether and 1.2 kg (0.3 g/g) of Raney nickel to a 100 L reaction kettle, introduce hydrogen until the system pressure is 0.8±0.1 MPa, regulate the pH of the system to 3±0.5 with concentrated sulfuric acid, add an acetonitrile solution containing 3.9 kg (1 eq) of (4S,5S)-2,2,5-trimethyl-5-(2-azidoacetyl)-1,3-dioxolane
react at 27±5° C. for 8.5 hours, perform suction filtration and concentration to obtain 2.3 kg of (3S,4S)-1-amino-3,4-dihydroxy-2-pentanone
with a yield of 89%;
Step 8: add 23 L (10 ml/g) of propanol, 6.9 L (3 ml/g) of pure water, 0.9 kg of (0.3 eq) of potassium iodide, 4.8 kg (1.2 eq) of compound A (2-amino-6-chloro-5-nitro-3H-pyrimidin-4-one), 2.3 kg (1 eq) of (3S,4S)-1-amino-3,4-dihydroxy-2-pentanone
and 10.5 kg (6 eq) of diisopropylamine to a 50 L reaction bottle, react the system for 7 hours while preserving the temperature at 72±5° C., then add a potassium dihydrogen phosphate-dipotassium phosphate aqueous solution to regulate the pH of the system to 7.5±0.5; and filter the system to obtain 2.5 kg of 2-acetylamino-5-nitro-6-((3S,4S)-3,3-dihydroxy-2-oxo-pentylamino)-pyrimidin-4-one
with a yield of 44%;
Step 9: add 1.25 kg (1 eq) of 2-acetylamino-5-nitro-6((3S,4S)-3,3-dihydroxy-2-oxo-pentylamino)-pyrimidin-4-one
50 L (40 ml/g) of ethanol and 0.5 kg (0.4 g/g) of 10% palladium on carbon to a 100 L autoclave, introduce hydrogen until the reaction system pressure is 0.8±0.05 MPa, control the temperature of the system at 27±5° C. and the pressure at 0.8±0.05 MPa, react for 24 hours, filter the system, and regulate the pH to 11±0.5 to obtain an ethanol solution containing 1.1 kg of acetylamino-7,8-dihydropteridine
which is used directly in the next step;
Step 10: add 0.44 kg (0.4 g/g) of palladium 10% on carbon in the presence of the ethanol solution containing 1.1 kg of acetylamino-7,8-dihydropteridine
obtained in Step 9, introduce hydrogen until the pressure of the reaction kettle is 0.8±0.05 MPa, control the temperature of the system at 25±5° C. and the pressure at 0.8±0.05 MPa, react for 80 hours, after reacting thoroughly, perform quenching in 20 kg (8 eq) of dilute hydrochloric acid having a concentration of 15%, and perform suction filtration and drying to the system to obtain a target product, i.e. a crude product of sapropterin dihydrochloride
recrystallize and purify the crude product by 21.4 L (20 ml/g) of ethanol at 35±5° C. to obtain 0.4 kg of a pure product, with a yield of 46.2%, a purity of 98.5% and an enantiomeric excess of 99.2%.
Embodiment 6: main raw material:
and X═N
Step 1: add 522 kg (0.05 eq) of a tetrahydrofuran solution containing a samarium catalyst having a concentration of 2%, 9.1 kg (0.05 eq) of (R)-(+)-1,1′-bi-2-naphthol, 8.9 kg (0.05 eq) of triphenylphosphine oxide, and 567 kg (7 kg/kg) of a 4 A molecular sieve to a 3000 L reaction kettle, after stirring uniformly, control the system temperature at 8±5° C., add 57.4 kg (0.eq) of a tert-butyl hydroperoxide toluene solution having a concentration of 50%, add 81.1 kg (1 eq) of (E)-N-isopropylbut-2-enamide
to the system after adding the tert-butyl hydroperoxide toluene solution, react for 34 hours while preserving the temperature, add 6.1 kg (0.05 eq) of citric acid to the system to stop the reaction, and perform centrifugation, concentration and rectification to the system to obtain 56.1 kg of (2S,3R)-2,3-epoxy-diisopropylamido butyrate
with a yield of 61.5%;
Step 2: add 11.4 kg (0.5 eq) of acetone, and 8.8 kg (0.1 eq) of zinc bromide to a 500 L enamel vessel, control the temperature at 20±5° C., add 56.1 kg (1 eq) of (2S,3R)-2,3-epoxy-diisopropylamido butyrate
react for 8.5 hours while preserving the temperature, add 329 kg (2 eq) of a sodium bicarbonate aqueous solution having a concentration of 10%, and perform liquid separation, extraction and concentration to the system to obtain 64.7 kg of (4S,5S)-2,2,5-trimethyl-2,3-acetonide-diisopropylamido butyrate
with a yield of 82%;
Step 3: add 259 L (4 ml/g) of tetrahydrofuran, and 64.7 kg (1 eq) of (4S,5S)-2,2,5-trimethyl-2,3-acetonide-diisopropylamido butyrate
to a 1000 L reaction kettle, increase the temperature to 27±5° C., add 2.9 kg (0.5 eq) of pure water and 29.9 kg (0.5 eq) of a methanol solution of sodium methoxide having a concentration of 29.9%, react for 4 hours while preserving the temperature, perform centrifugation, dissolve a filter cake in 194 L (3 ml/g) of tetrahydrofuran, add 39 kg (1 eq) of L-α-phenylethylamine, preserve the temperature at 18±5° C. for 3.5 hours, and perform centrifugation and drying to obtain 54.3 kg of 1-phenyltehanamine (4S,5S)-2,2,5-trimethyl-1,3-dioxolane-4-carboxylate
with a yield of 60%;
Step 4: add 30 L (3 ml/g) of 2-methyltetrahydrofuran, 10 kg (1 eq) of 1-phenyltehanamine (4S,5S)-2,2,5-trimethyl-1,3-dioxolane-4-carboxylate
to a 72 L reaction bottle, then add a dilute phosphoric acid aqueous solution having a concentration of 10% to the system to regulate the pH at 1.5±0.5, control the temperature at −5±5° C., react for 1 hour, perform liquid separation to obtain an organic phase, add 3.7 kg of (0.8 eq) N,N-diisopropylethylamine to the organic phase, and concentrate the system to obtain 5.3 kg of (4S,5S)-2,2,5-trimethyl-1,3-dioxolan-4-methanoic acid
with a yield of 92.5%;
Step 5: add 42 L (8 ml/g) of 1,4-dioxane, 5.3 kg of 1,3-dioxolan-4-methanoic acid
and 8.5 kg (2 eq) of N,N-diisopropylethylamine to a 100 L reaction bottle, reduce the temperature to −10±5° C., add 4 kg (21.0 eq) of propyl chloroformate, react for 2 hours while preserving the temperature, introduce a diazomethane gas for 2 hours, add 12 kg (2 eq) of a hydrochloride ethanol solution having a concentration of 20%, react for 2 hours, add sodium hydroxide to regulate the pH value to 7.5±0.5, and perform extraction, liquid separation and concentration to obtain 5.1 kg of (4S,5S)-2,2,5-trimethyl-5-chloroacetyl-1,3-dioxolane
with a yield of 81%; Step 6: add 41 L (8 ml/g) of tetrahydrofuran, 5.1 kg of (4S,5S)-2,2,5-trimethyl-5-chloroacetyl-1,3-dioxolane
3.1 kg (1 eq) of azidotrimethylsilane, and 0.5 kg (0.1 eq) of sodium iodide to a 72 L bottle, react the system for 30 hours while preserving the temperature at 12±5° C., perform filtering and concentration to obtain an acetone solution containing 4.6 kg of (4S,5S)-2,2,5-trimethyl-5-(2-azidoacetyl)-1,3-dioxolane
with a yield of 87.5%;
Step 7: add 28 L (6 ml/g) of 1,4-dioxane and 0.23 kg (0.05 g/g) of palladium 10% on carbon to a 50 L reaction kettle, introduce hydrogen until the system pressure is 0.8±0.1 MPa, regulate the pH of the system to 3±0.5 with acetic acid, add an acetonitrile solution containing 4.6 kg (1 eq) of (4S,5S)-2,2,5-trimethyl-5-(2-azidoacetyl)-1,3-dioxolane
react at 27±5° C. for 8.5 hours, react for 8.5 hours, perform suction filtration and concentration to obtain 2.7 kg of (3S,4S)-1-amino-3,4-dihydroxy-2-pentanone
with a yield of 87.7%;
Step 8: add 16.3 L (6 ml/g) of isopropanol, 2.7 L (1 g/g) of pure water, 0.4 kg of (0.1 eq) of sodium iodide, 4.8 kg (1.0 eq) of compound A (2-amino-6-chloro-5-nitro-3H-pyrimidin-4-one), 2.7 kg (1 eq) of (3S,4S)-1-amino-3,4-dihydroxy-2-pentanone
and 8.7 kg (4 eq) of sodium carbonate to a 50 L reaction bottle, react the system for 7 hours while preserving the temperature at 45±5° C., then add an ammonium formate-ammonia aqueous solution to regulate the pH of the system to 6.5±0.5; and filter the system to obtain 2.85 kg of 2-acetylamino-5-nitro-6((3S,4S)-3,3-dihydroxy-2-oxo-pentylamino)-pyrimidin-4-one
with a yield of 42.5%;
Step 9: add 2 kg (1 eq) of 2-acetylamino-5-nitro-6-((3S,4S)-3,3-dihydroxy-2-oxo-pentylamino)-pyrimidin-4-one
60 L (30 ml/g) of ethanol and 0.2 kg (0.1 g/g) of platinum dioxide to a 100 L autoclave, introduce hydrogen until the reaction system pressure is 0.6±0.05 MPa, control the temperature of the system at 20±5° C. and the pressure at 0.6±0.05 MPa, react for 20 hours, filter the system, and regulate the pH to 11±0.5 to obtain an ethanol solution containing 1.7 kg of acetylamino-7,8-dihydropteridine
which is used directly in the next step;
Step 10: add 0.2 kg (0.1 g/g) of platinum dioxide in the presence of the ethanol solution containing 1.7 kg of acetylamino-7,8-dihydropteridine
obtained in Step 9, introduce hydrogen until the pressure of the reaction kettle is 0.6±0.05 MPa, control the temperature of the system at 15±5° C. and the pressure at 0.6±0.05 MPa, react for 75 hours, after reacting thoroughly, perform quenching in 30 kg (5 eq) of dilute hydrochloric acid having a concentration of 10%, and perform suction filtration and drying to the system to obtain a target product, i.e. a crude product of sapropterin dihydrochloride
recrystallize and purify the crude product by 17 L (10 ml/g) of butanone at 15±5° C. to obtain 0.6 kg of a pure product, with a yield of 43%, a purity of 98.4% and an enantiomeric excess of 98.9%.
Thus, it can be seen that synthesis of a sapropterin dihydrochloride compound and an intermediate thereof disclosed in a method of the present disclosure can obtain a target product with a high purity, a high enantiomeric excess, and a high yield. The synthesis method uses readily-available raw materials, significantly reduces a synthesis route of sapropterin dihydrochloride. The technological conditions are stable, and there is less pollution in the whole operation process, hence providing an effective scheme for mass industrial production of sapropterin dihydrochloride.
The above are only preferred embodiments of the present disclosure and should not be used to limit the present disclosure. For those skilled in the art, the present disclosure may have various modifications and changes. Any modifications, equivalent replacements, improvements and the like within the spirit and principle of the present disclosure shall fall within the scope of protection of the present disclosure.
///////
References
- ^ Jump up to:a b “Sapropterin (Kuvan) Use During Pregnancy”. Drugs.com. 17 May 2019. Retrieved 4 March 2020.
- ^ “Sapropterin”. Drugs.com. 28 February 2020. Retrieved 4 March 2020.
- ^ “International Non-proprietary Names for Pharmaceutical Substances (INN)”. Fimea. Retrieved 4 March 2020.
- ^ Jump up to:a b Kappock TJ, Caradonna JP (November 1996). “Pterin-Dependent Amino Acid Hydroxylases”. Chemical Reviews. 96 (7): 2659–2756. doi:10.1021/CR9402034. PMID 11848840.
- ^ Całka J (2006). “The role of nitric oxide in the hypothalamic control of LHRH and oxytocin release, sexual behavior and aging of the LHRH and oxytocin neurons”. Folia Histochemica et Cytobiologica. 44 (1): 3–12. PMID 16584085.
- ^ Bhagavan, N. V. (2015). Essentials of Medical Biochemistry With Clinical Cases, 2nd Edition. USA: Elsevier. p. 256. ISBN 978-0-12-416687-5.
- ^ Schaub J, Däumling S, Curtius HC, Niederwieser A, Bartholomé K, Viscontini M, et al. (August 1978). “Tetrahydrobiopterin therapy of atypical phenylketonuria due to defective dihydrobiopterin biosynthesis”. Archives of Disease in Childhood. 53 (8): 674–6. doi:10.1136/adc.53.8.674. PMC 1545051. PMID 708106.
- ^ Jump up to:a b “Kuvan- sapropterin dihydrochloride tablet Kuvan- sapropterin dihydrochloride powder, for solution Kuvan- sapropterin dihydrochloride powder, for solution”. DailyMed. 13 December 2019. Retrieved 4 March 2020.
- ^ Jump up to:a b c “Kuvan EPAR”. European Medicines Agency (EMA). 4 March 2020. Retrieved 4 March 2020.
- ^ “Drug Approval Package: Kuvan (Sapropterin Dihydrochloride) NDA #022181”. U.S. Food and Drug Administration (FDA). 24 March 2008. Retrieved 4 March 2020. Lay summary (PDF).
- ^ Jump up to:a b c d e “Kuvan (sapropterin dihydrochloride) Tablets and Powder for Oral Solution for PKU”. BioMarin. Retrieved 4 March 2020.
- ^ “Drug Approval Package: Kuvan Powder for Oral Solution (Sapropterin Dihydrochloride) NDA #205065”. U.S. Food and Drug Administration (FDA). 28 February 2014. Retrieved 4 March 2020. Lay summary (PDF).
- ^ Herper M (28 July 2016). “How Focusing On Obscure Diseases Made BioMarin A $15 Billion Company”. Forbes. Retrieved 9 October 2017.
- ^ “BioMarin Announces Kuvan (sapropterin dihydrochloride) Patent Challenge Settlement”. BioMarin Pharmaceutical Inc. 13 April 2017. Retrieved 9 October 2017 – via PR Newswire.
- ^ “Tetrahydrobiopterin Deficiency”. National Organization for Rare Disorders (NORD). Retrieved 9 October 2017.
- ^ “What are common treatments for phenylketonuria (PKU)?”. NICHD. 23 August 2013. Retrieved 12 September 2016.
- ^ Camp KM, Parisi MA, Acosta PB, Berry GT, Bilder DA, Blau N, et al. (June 2014). “Phenylketonuria Scientific Review Conference: state of the science and future research needs”. Molecular Genetics and Metabolism. 112 (2): 87–122. doi:10.1016/j.ymgme.2014.02.013. PMID 24667081.
- ^ Jump up to:a b Haberfeld, H, ed. (1 March 2017). Austria-Codex (in German). Vienna: Österreichischer Apothekerverlag. Kuvan 100 mg-Tabletten.
- ^ “Genetics Home Reference: GCH1”. National Institutes of Health.
- ^ Wu G, Meininger CJ (2009). “Nitric oxide and vascular insulin resistance”. BioFactors. 35(1): 21–7. doi:10.1002/biof.3. PMID 19319842. S2CID 29828656.
- ^ Kaufman S (February 1958). “A new cofactor required for the enzymatic conversion of phenylalanine to tyrosine”. The Journal of Biological Chemistry. 230 (2): 931–9. PMID 13525410.
- ^ Thöny B, Auerbach G, Blau N (April 2000). “Tetrahydrobiopterin biosynthesis, regeneration and functions”. The Biochemical Journal. 347 Pt 1: 1–16. doi:10.1042/0264-6021:3470001. PMC 1220924. PMID 10727395.
- ^ Kuzkaya N, Weissmann N, Harrison DG, Dikalov S (June 2003). “Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase”. The Journal of Biological Chemistry. 278 (25): 22546–54. doi:10.1074/jbc.M302227200. PMID 12692136.
- ^ Muller-Delp JM (November 2009). “Ascorbic acid and tetrahydrobiopterin: looking beyond nitric oxide bioavailability”. Cardiovascular Research. 84 (2): 178–9. doi:10.1093/cvr/cvp307. PMID 19744948.
- ^ Gori T, Burstein JM, Ahmed S, Miner SE, Al-Hesayen A, Kelly S, Parker JD (September 2001). “Folic acid prevents nitroglycerin-induced nitric oxide synthase dysfunction and nitrate tolerance: a human in vivo study”. Circulation. 104 (10): 1119–23. doi:10.1161/hc3501.095358. PMID 11535566.
- ^ “Search results for Kuvan”. ClinicalTrials.gov. U.S. National Library of Medicine.
- ^ “BioMarin Initiates Phase 3b Study to Evaluate the Effects of Kuvan on Neurophychiatric Symptoms in Subjects with PKU”. BioMarin Pharmaceutical Inc. 17 August 2010.
- ^ Burton B, Grant M, Feigenbaum A, Singh R, Hendren R, Siriwardena K, et al. (March 2015). “A randomized, placebo-controlled, double-blind study of sapropterin to treat ADHD symptoms and executive function impairment in children and adults with sapropterin-responsive phenylketonuria”. Molecular Genetics and Metabolism. 114 (3): 415–24. doi:10.1016/j.ymgme.2014.11.011. PMID 25533024.
- ^ Fernell E, Watanabe Y, Adolfsson I, Tani Y, Bergström M, Hartvig P, et al. (May 1997). “Possible effects of tetrahydrobiopterin treatment in six children with autism–clinical and positron emission tomography data: a pilot study”. Developmental Medicine and Child Neurology. 39 (5): 313–8. doi:10.1111/j.1469-8749.1997.tb07437.x. PMID 9236697. S2CID 12761124.
- ^ Frye RE, Huffman LC, Elliott GR (July 2010). “Tetrahydrobiopterin as a novel therapeutic intervention for autism”. Neurotherapeutics. 7 (3): 241–9. doi:10.1016/j.nurt.2010.05.004. PMC 2908599. PMID 20643376.
- ^ Channon KM (November 2004). “Tetrahydrobiopterin: regulator of endothelial nitric oxide synthase in vascular disease”. Trends in Cardiovascular Medicine. 14 (8): 323–7. doi:10.1016/j.tcm.2004.10.003. PMID 15596110.
- ^ Alp NJ, Mussa S, Khoo J, Cai S, Guzik T, Jefferson A, et al. (September 2003). “Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression”. The Journal of Clinical Investigation. 112 (5): 725–35. doi:10.1172/JCI17786. PMC 182196. PMID 12952921.
- ^ Khoo JP, Zhao L, Alp NJ, Bendall JK, Nicoli T, Rockett K, et al. (April 2005). “Pivotal role for endothelial tetrahydrobiopterin in pulmonary hypertension”. Circulation. 111 (16): 2126–33. doi:10.1161/01.CIR.0000162470.26840.89. PMID 15824200.
- ^ Cunnington C, Van Assche T, Shirodaria C, Kylintireas I, Lindsay AC, Lee JM, et al. (March 2012). “Systemic and vascular oxidation limits the efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease”. Circulation. 125 (11): 1356–66. doi:10.1161/CIRCULATIONAHA.111.038919. PMC 5238935. PMID 22315282.
- ^ Romanowicz J, Leonetti C, Dhari Z, Korotcova L, Ramachandra SD, Saric N, et al. (August 2019). “Treatment With Tetrahydrobiopterin Improves White Matter Maturation in a Mouse Model for Prenatal Hypoxia in Congenital Heart Disease”. Journal of the American Heart Association. 8 (15): e012711. doi:10.1161/JAHA.119.012711. PMC 6761654. PMID 31331224.
- ^ Kraft VA, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. (January 2020). “GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling”. ACS Central Science. 6 (1): 41–53. doi:10.1021/acscentsci.9b01063. PMC 6978838. PMID 31989025.
Further reading
- “Clinical Review Report: Sapropterin dihydrochloride (Kuvan)”. CADTH Common Drug Reviews. Ottawa, Canada: Canadian Agency for Drugs and Technologies in Health (CADTH). September 2017. Bookshelf ID: NBK533813.
- Blau N (June 2016). “Genetics of Phenylketonuria: Then and Now”. Human Mutation. 37 (6): 508–15. doi:10.1002/humu.22980. PMID 26919687.
- Dubois EA, Cohen AF (June 2010). “Sapropterin”. British Journal of Clinical Pharmacology. 69 (6): 576–7. doi:10.1111/j.1365-2125.2010.03643.x. PMC 2883749. PMID 20565448.
- Muntau AC, Adams DJ, Bélanger-Quintana A, Bushueva TV, Cerone R, Chien YH, et al. (May 2019). “International best practice for the evaluation of responsiveness to sapropterin dihydrochloride in patients with phenylketonuria”. Molecular Genetics and Metabolism. 127 (1): 1–11. doi:10.1016/j.ymgme.2019.04.004. PMID 31103398.
- Qu J, Yang T, Wang E, Li M, Chen C, Ma L, et al. (May 2019). “Efficacy and safety of sapropterin dihydrochloride in patients with phenylketonuria: A meta-analysis of randomized controlled trials”. British Journal of Clinical Pharmacology. 85 (5): 893–899. doi:10.1111/bcp.13886. PMC 6475685. PMID 30720885.
- van Wegberg AM, MacDonald A, Ahring K, Bélanger-Quintana A, Blau N, Bosch AM, et al. (October 2017). “The complete European guidelines on phenylketonuria: diagnosis and treatment”. Orphanet Journal of Rare Diseases. 12 (1): 162. doi:10.1186/s13023-017-0685-2. PMC 5639803. PMID 29025426.
External links
- “Sapropterin”. Drug Information Portal. U.S. National Library of Medicine.
Tetrahydrobiopterin
INN: sapropterin
![(6R)-Tetrahydrobiopterin structure.png]() |
| Clinical data |
| Trade names |
Kuvan, Biopten |
| Other names |
Sapropterin hydrochloride (JAN JP), Sapropterin dihydrochloride (USAN US) |
| AHFS/Drugs.com |
Monograph |
| MedlinePlus |
a608020 |
| License data |
|
Pregnancy
category |
- AU: B1[1]
- US: C (Risk not ruled out)[1]
|
Routes of
administration |
By mouth |
| ATC code |
|
| Legal status |
| Legal status |
- AU: S4 (Prescription only)
- CA: ℞-only
- US: ℞-only
- In general: ℞ (Prescription only)
|
| Pharmacokinetic data |
| Elimination half-life |
4 hours (healthy adults)
6–7 hours (PKU patients) |
| Identifiers |
|
|
| CAS Number |
|
| PubChem CID |
|
| IUPHAR/BPS |
|
| DrugBank |
|
| ChemSpider |
|
| UNII |
|
| KEGG |
|
| ChEBI |
|
| ChEMBL |
|
| PDB ligand |
|
| CompTox Dashboard (EPA) |
|
| ECHA InfoCard |
100.164.121 ![Edit this at Wikidata]() |
| Chemical and physical data |
| Formula |
C9H15N5O3 |
| Molar mass |
241.251 g·mol−1 |
| 3D model (JSmol) |
|
-
CC(C(C1CNC2=C(N1)C(=O)N=C(N2)N)O)O
|
-
InChI=1S/C9H15N5O3/c1-3(15)6(16)4-2-11-7-5(12-4)8(17)14-9(10)13-7/h3-4,6,12,15-16H,2H2,1H3,(H4,10,11,13,14,17)/t3-,4+,6-/m0/s1 ![☑]()
-
Key:FNKQXYHWGSIFBK-RPDRRWSUSA-N ![☑]()
|
////////Sapropterin, сапроптерин , سابروبتيرين , 沙丙蝶呤 , Tetrahydrobiopterin,
![Sapropterin Tablets - FDA prescribing information, side effects and uses]()









.jpg)




















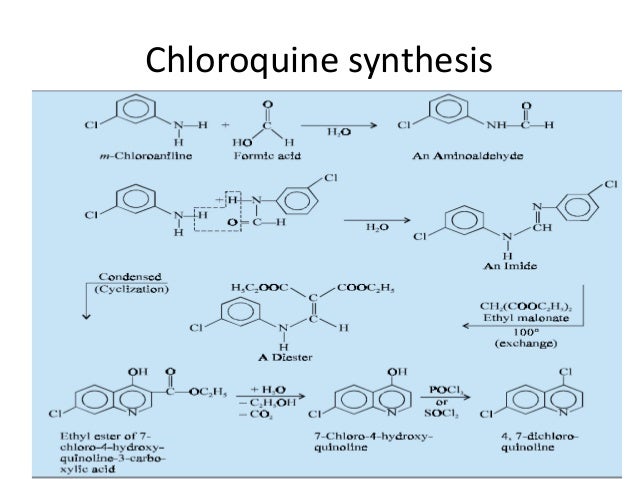
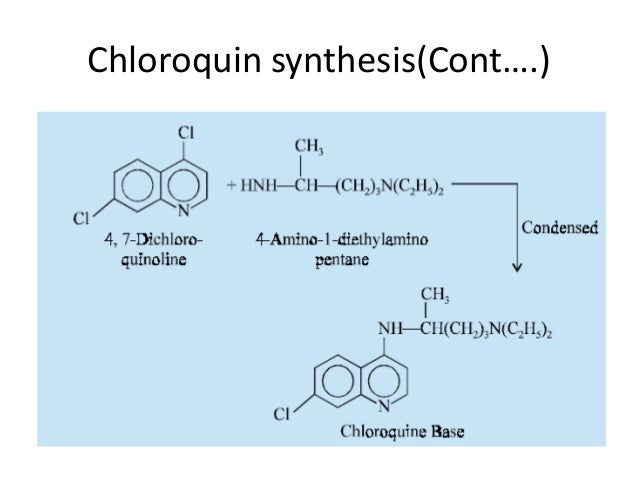
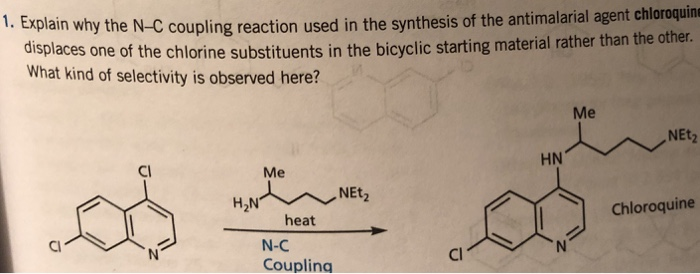
 Elevated occurrence of chloroquine- or multi-resistant malaria
Elevated occurrence of chloroquine- or multi-resistant malaria

























![(1R,5S,6R,7R,10R,11R,14R,15S,20R,21R)-21-[(2R)-2-Amino-2,3,3-trimethylbutoxy]-20-(5-carbamoyl-1,2,4-triazol-1-yl)-5,7,10,15-tetramethyl-7-[(2R)-3-methylbutan-2-yl]-17-oxapentacyclo[13.3.3.01,14.02,11.05,10]henicos-2-ene-6-carboxylic acid.png](http://pubchem.ncbi.nlm.nih.gov/image/imgsrv.fcgi?cid=59312774&t=l)